Секвенируй это
Ликбез по технологиям чтения ДНК
Каждые два года количество транзисторов, размещаемых на интегральной микросхеме, увеличивается вдвое. Так звучит каноническая версия закона Мура, – эмпирического наблюдения одного из основателей Intel, которое известно каждому, интересующемуся новостями технологий. Как драйвер невероятного успеха IT-индустрии закон Мура давно стал, пожалуй, самым наглядным маркером прогресса. Однако есть области технологий, где даже эта «икона сингулярности» выглядит довольно бледно. Речь идет о технологиях чтения ДНК.
Первый человеческий геном, полученный в рамках десятилетнего международного проекта, обошелся примерно в три миллиарда долларов — это стоимость всей программы, включающая многочисленные научные исследования, который приходилось проводить по ходу работы. На момент окончания проекта (черновая версия генома была опубликована в 2001 году) стоимость прочтения еще одного генома сравнимого размера оценивалась приблизительно в 100 миллионов долларов. Нетрудно подсчитать, что сейчас, 14 лет спустя, если бы закон Мура действовал в биотехе, стоимость секвенирования составила бы около 750 тысяч долларов. На самом деле по состоянию на 2015 год стоимость чтения полного человеческого генома составляет около пяти тысяч долларов, а цена генотипирования — анализа только избранных, «ключевых» участков генома — опустилась уже до сотни долларов. Даже с учетом подготовки проб и логистики оба варианта процесса занимают уже не годы, а примерно несколько недель.

Закон Мура и динамика стоимости секвенирования генома.
На второй вопрос ответить проще: «взрыв» геномной революции действительно произошел, но до нас, обычных потребителей, пока не дошла его «взрывная волна». Геномные данные уже давно стали радикально доступнее ученым и фармкомпаниям, но в жизнь самих их обладателей геномные данные стали выходить только в последние несколько лет. А вот первый вопрос — как и почему это произошло, как менялась технология секвенирования и как сегодня ученые читают ДНК — требует отдельного разбора.
Для начала нужно определится с тем, что такое секвенирование. Секвенированием (от слова sequence — последовательность) называют определение порядка элементраных единиц мономеров в полимере. Причем эти полимером может быть не только ДНК или РНК, но и, например, белок или даже полисахарид. Сам термин «секвенирование» возник в тот момент, когда стало понятно, что в биологии свойства и функции полимеров не могут определяться просто их составом, как привыкли думать химики: слишком похож оказался этот состав у совершенно разных с функциональной точки зрения молекул. А если дело не в составе, значит ключевую роль играет именно последовательность мономеров — идея, которая кажется тривиальной сейчас, но в 40-е годы прошлого века была совершенно новой.

Приоритет в открытии этой ключевой для биологии концепции принадлежит британскому ученому Фредерику Сенгеру. «Папа секвенирования» родился в 1918 году в семье врача и прожил очень длинную и исключительно плодотворную научную жизнь. Единственный в мире дважды нобелевский лауреат по химии (1958 и 1980 годов) он в полной мере успел застать геномную революцию, созданную прежде всего своими же руками.
Однако объектом первого в мире секвенирования, которое в конце 40-х провел Сенгер, была вовсе не ДНК или РНК. Это был инсулин – единственный в то время пептид, доступный в более-менее чистом виде в достаточных количествах. То, какие аминокислоты входят в состав инсулина было к тому моменту известно, но ученые полагали, что пропорции этих аминокислот в инсулине приблизительны, да и не слишком важны — предполагалось, что при создании белков жизнь руководствуется «аналоговым» подходом, «насыпая» аминокислоты в разные белки на глазок.
Именно Сенгер показал, что это не так. Ему удалось найти реагент, который избирательно взаимодействует только с одной аминокислотой из всей полипептидной цепочки: той, которая находится в самом начале пептида, и у которой, из-за этого, есть уникальная химическая группа. И если для исходного полипептида такая аминокислота одна, то после частичного разрезания концевой может стать любая из аминокислот, а значит все их можно идентифицировать. Путем разбивания инсулина на мелкие фрагменты и определения концевых аминокислот Сенгеру удалось (всего-то за восемь лет кропотливой работы) собрать полный «пазл» мономеров, и определить, таким образом, точную структуру инсулина. За эту работу Сенгер спустя всего шесть лет после последней публикации получил Нобелевскую премию по химии.
Однако после успеха с инсулином никто — ни Сенгер, ни его конкуренты, даже не пытались подступиться к секвенированию ДНК. «Самая главная молекула» выглядела слишком огромной и устрашающей для того, чтобы можно было попытаться прочитать ее последовательность. Кроме того, в каждой клетке, как известно, содержится всего одна или две копии геномной ДНК, а значит получить достаточное количество одинаковых молекул (чего требует метод), довольно сложно.

Тактической целью стала другая нуклеиновая кислота, РНК. Точнее говоря, ее отдельная разновидность, транспортная РНК, которая в клетке используется в качестве адаптера, «подносящего» аминокислоты при синтезе белка. Ее длина составляет всего 70-80 нуклеотидов, а число копий на клетку достигает сотен тысяч штук. Для секвенирования РНК Сенгер применил ту же общую стратегию: мечение концевого мономера с последующим частичным разрушением молекулы на множество фрагментов. Однако тут удача ему изменила. Роберт Холли с соавторами из Корнельского университета смог опубликовать последовательность одной из тРНК дрожжей уже в 1965 году, именно Холли стал первым человеком, определившим последовательность какой-либо нуклеиновой кислоты. И хотя всего три года спустя группе Сенгера удалось секвенировать РНК почти вдвое длиннее (это была одна из тех молекул, что входят в состав рибосомы), по-настоящему отыграться за это поражение ему удалось только спустя десять лет.
Метод секвенирования ДНК, несущий сейчас имя Сенгера, был опубликован в 1977 году. На протяжении более 30 лет, вплоть до середины нулевых годов, он оставался главным способом определения последовательности любой нуклеиновой кислоты: именно этим методом (с незначительными модификациями) был прочитан геном человека. И до сих пор секвенирование по Сенгеру является самым точным методом, к которому обращаются при необходимости проверить результаты секвенирования нового поколения.
Идея, лежащая в основе секвенирования по Сенгеру, настолько проста и изящна, что на ней хочется остановится подробнее. До сих пор, как мы видели, все методы чтения последовательности были основаны, условно говоря, на разрушении: на получении чистого вещества, его фрагментировании и восстановлении «пазла» из получившихся фрагментов. Сенгер решил действовать противоположным образом: читать ДНК не фрагментированием, а синтезом, причем использовать для этого природный фермент, ДНК полимеразу — ту самую молекулярную машину, которая удваивает ДНК перед делением клетки.
Процедура проводится следующим образом: фрагмент ДНК, помеченный с одного из концов радиоактивным изотопом, разделяют на четыре пробирки. В каждую из них добавляют реактивы, необходимые для синтеза новой ДНК, в том числе одиночные «буквы», которые полимераза должна будет связать в новую нить — в точном соответствии с исходной матрицей. Однако помимо обычных «букв» к раствору добавляется некоторое небольшое число специально «испорченных» — таких, после которых невозможно присоединить следующую «букву» (они просто лишены соответствующего места для связи).
В результате в конце синтеза в каждой пробирке появляется набор ДНК разной длины, причем каждая из молекул несет радиоактивную метку в начале и испорченную «букву» на конце. Поскольку в каждую из четырех пробирок мы кладем только один вид испорченных оснований, мы знаем, какой буквой кончаются все фрагменты ДНК в данной пробирке. Теперь достаточно разогнать содержимое всех четырех пробирок в геле, разделяющем ДНК по длине и приложить к нему фотопленку, которая зафиксирует скопления радиоактивности. На пленке появится «лестница» из ступенек, каждая из которых будет соответствовать одной букве в последовательности. Поднимаясь по лестнице, мы прочтем всю последовательность исходной ДНК.

Метод Сенгера оказался исключительно надежным и удобным для чтения последовательностей. Возможно, именно благодаря тому, что он полагался на «выдрессированные» в ходе миллиардов лет эволюции природные ферменты, а не синтетические реактивы. Именно с помощью этого метода (точнее, с помощью его непосредственного варианта-предшественника) Сенгеру удалось прочитать первый в истории полный геном отдельного организма — геном бактериофага ϕX174, содержащий всего 5386 оснований (кстати, этот же фаг в 2003 году стал первым организмом, геном которого был полностью синтезирован искусственно).
Уже несколько лет спустя, в середине 80-х, когда инкрементные улучшения постепенно увеличивали скорость и мощность секвенирования, стали появляться полные геномы все более сложных вирусов и ученые впервые заговорили о возможном секвенировании геномов высших организмов, в том числе и человека.
Подобраться к этой задаче стало возможно тогда, когда появились методы молекулярного клонирования, позволявшие вырезать из генома отдельные фрагменты, затем вставлять их в модельные организмы (кишечную палочку или дрожжи), а потом постепенно, по кусочку, секвенировать – тем же самым методом Сенгера.
В 1990 году стартовала международная программа «Геном человека», в которой каждому коллективу из Америки, Европы и Японии были выделены отдельные участки предварительно размеченного генома для секвенирования. К этому моменту несколько биотехнологических компаний, прежде всего Applied Biosciences, научились автоматизировать процессы секвенирования по Сенгеру. Сначала секвенаторы могли самостоятельно только читать «лестницы» на фотопленке, превращая их в буквенную последовательность, а затем сам процесс разделения фрагментов ДНК удалось перенести из геля (который нужно было каждый раз заливать заново) в тонкий капилляр. Радиоактивные метки заменили флюоресцентными, и это позволило читать последовательность прямо во время прохода сквозь капилляр: четыре цвета — четыре разные буквы.
Одним из первых, кто осознал возможности автоматизации секвенирования, был Крейг Вентер — будущий одиозный зачинатель «гонки геномов», решивший опередить в прочтении генома человека всю международную коллаборацию (а также автор первого живого организма с полностью синтетической ДНК). В 1992 году, спустя два года после старта проекта «Геном человека», Вентер организовал собственный институт с броской аббревиатурой TIGR (The Institute for Genomic Research), в котором секвенирование ДНК было впервые поставлено на поток. В 1995 году группе Вентера удалось получить первый геном полноценного клеточного организма, точнее даже двух: бактерий Haemophilus influenzae и Mycoplasma genitalium.

С чисто биологической точки зрения принципиальным было то, что речь шла именно о полноценных клеточных геномах: вирусы живут только в клетках и в своей жизни почти во всем полагаются на клеточные системы, кодируемые не собственным геномом, а геномом клетки. И только последние содержат полную необходимую для жизни информацию и поэтому гораздо интереснее вирусных геномов.
С технической точки зрения новаторство Вентера заключалось в том, что он впервые применил радикальный подход к секвенированию генома. Как уже говорилось, до сих пор ученые работали с геномом «по кусочкам», что позволяло читать ДНК самых сложных организмов но и требовало огромных затрат времени на клонирование. Вентер стал пионером так называемого «метода дробовика», когда весь геном целиком нарезается на множество коротких пересекающихся фрагментов, которые прочитываются, а затем собираются снова: снизу вверх.
Такой подход существенно упрощает все стадии, требующие участия человека и хорошо подходит для автоматизации. Однако у него есть два принципиальных недостатка. Во-первых, сборка генома из миллионов и миллионов коротких фрагментов требует огромных вычислительных ресурсов, подразумевает создание принципиально новых математических алгоритмов и требует многократного покрытия (например, чтобы собрать геном длиной 100 нуклеотидов вам нужно прочитать фрагментов общей длиной в 1000 — в этом случае говорят про десятикратное покрытие).
А во-вторых, работает этот метод по принципу «пока не сделано всё — не сделано ничего». Именно благодаря этой особенности наблюдать за гонкой геномов было особенно интересно: если бы качества и количество «сырых» последовательностей Вентеру немного не хватило, вся авантюра c секвенированием оказалась бы пустой тратой времени. Этого, однако, не случилось. Оба генома — и полученный в ходе международного проекта и собранный частной компанией Вентера были опубликованы в двух выпусках Nature и Science, вышедших на одной неделе в феврале 2001 года.
Услышав про проект «Геном человека» многие спрашивают: — а какого именно? Кем был тот человек, на прочтение генома которого было потрачено столько денег и усилий ученых? И хотя ответ тривиален (никем он не был, это референтный геном, ДНК для которого была получена у нескольких анонимных доноров) такой вопрос бьет в самую точку. Никто из нас не является обладателем консенсусного генома, в ДНК каждого существует огромное количество уникальных отличий, и именно эти отличия делают нас теми, кто мы есть. Поэтому в первый же день после публикации референтного генома человека стало ясно, что главной целью всех последующих исследований в человеческой геномике станут индивидуальные отличия.
Есть определенная ирония в том, что первыми людьми, персональный геном которых был прочитан, стали два главных соперника «гонке геномов»: первооткрыватель структуры ДНК Джеймс Уотсон и уже знакомый нам Крейг Вентер. По сравнению с референтным геномом у каждого из них было обнаружено около трех миллионов индивидуальных полиморфизмов — однобуквенных замен, которые отличаются от человека к человеку. Стоимость секвенирования обоих индивидуальных геномов к моменту их прочтения в 2007 году составила около миллиона долларов. И это, конечно существенно ниже, чем 100 миллионов в 2001 году, но все равно немало: с такими ценами рассчитывать на прочтение геномов сотен или тысяч человек или предлагать такую услугу обычным людям было бы странно. Однако, к счастью, как раз в тот момент, когда референтный геном был создан, созрела технология, позволяющая «вылавливать» индивидуальные отличия в геномах принципиально проще и дешевле обычного секвенирования. Речь идет о технологии ДНК-микрочипов.

ДНК-микрочип — это небольшая пластинка, на которой с помощью технологии, напоминающей фотолитографию, закреплены короткие одноцепочечные фрагменты ДНК. При инкубации с раствором образца две молекулы ДНК — одна на чипе, другая в образце — могут образовать прочную пару. Если все молекулы образца предварительно пометить флюоресцентным маркером, то после инкубации мы увидим на чипе светящиеся точки в тех местах, где образовались прочные пары. И если это произошло, значит последовательности фрагментов ДНК в образце точно совпали с последовательностями на чипе — а их мы знали заранее.
Установив, какие в популяции существуют индивидуальные особенности, мы можем создать микрочип с сотнями тысяч различных полиморфизмов. Это позволит получать информацию о наличии тех или иных «однобуквенных» замен в вашей ДНК всего за несколько часов. Формально, такая процедура не является секвенированием, но она позволяет читать последовательности ДНК, варианты который мы уже знаем. Использовать микрочипы для чтения совершенно новых последовательностей нельзя (хотя работы в этом направлении ведутся), но когда речь идет о персональной геномике, этого и не требуется: ДНК разных людей, как известно, совпадают на 99 процентов. С помощью современных микрочипов можно прочитать около миллиона известных полиморфизмов, то есть примерно одну треть от того их количества, которое присутствует в геноме.
ДНК-микрочипы стали появляться в научных лабораториях в 90-х, а в середине 2000-х появились первые компании, предлагающие анализ персонального генома на их основе. Небезызвестная 23andMe, основанная бывшей женой Сергея Брина, как раз была одной из первых таких компаний. Сейчас у компании Энн Вожитски появилось множество конкурентов, причем, как в мире, так и в России.
Однако сегодня технологиям генотипирования наступают на пятки (и по скорости, и по стоимости) так называемые методы секвенирования нового поколения. Именно их появление обвалило стоимость процедуры с миллионов до тысяч долларов. Это методы очень разные, и обо всех них рассказать не получится.
Немного остановится можно, пожалуй, только на так называемом пиросеквенировании — методе, который основан на гидролизе пирофосфата. При соединении нуклеотидов друг с другом в цепочку ДНК в раствор всегда выбрасывается это соединение — высокоэнергетичный фрагмент нуклеотида, который затем бесследно разрушается и своей «гибелью» обеспечивает однонаправленность реакции синтеза. В середине 2000-х многие научные группы независимо заметили, что разрушение пирофосфата уместно использовать при секвенировании: его можно «скармливать» специальному ферменту, который умеет превращать энергию связи пирофосфата в импульс света. Тогда, по наличию или отсутствию вспышки можно будет судить — прошла ли реакция присоединения нуклеотида к цепочке или нет. Когда на матрице ДНК образца синтезируется ее копия, вспышка означает наличие в нуклеиновой кислоте комплементарного основания.

Выглядит это так: ДНК режут на миллионы коротких фрагментов, наносят на микроскопические шарики, копируют их (так, чтобы на одном шарике были только идентичные копии одного фрагмента) и распределяют по микроскопическим ячейкам, сделанным в специальной подложке. После этого в ячейках начинается синхронная реакция. Сквозь подложку пропускают один вид нуклеотидов — если в ячейке при этом происходит вспышка, значит этот нуклеотид подходит для синтеза, значит на матрице находится комплементарный нуклеотид. Затем подложку отмывают от первого нуклеотида и подают второй — на этот раз загораются другие ячейки, те, в которых есть соответствующее комплементарное основание. Так, многократно промывая ячейки четырьмя нуклеотидами, биоинженеры читают последовательность ДНК по вспышкам в отдельных ячейках. Главная особенность этого и подобных методов — возможность проводить огромное количество параллельных реакций. И хотя точность реакции в каждой из ячеек невелика, огромное количество таких ячеек делает секвенирование очень быстрым и, следовательно, дешевым.
И все же пока полногеномное секвенирование не может сравняться по стоимости с генотипированием. Да, в исследовательских лабораториях технологии нового поколения уже вытесняют генотипирование из традиционных для этого метода задач (например, для анализа РНК и экспрессии генов). Но вот в персональной геномике дела обстоят иначе: особенности генома, которые заметны только при полном секвенировании и не видны микрочипам настолько редки, что настоящее секвенирование кажется стрельбой из пушки по воробьям.
В последние год-два на рынке полногеномного чтения ДНК наблюдается небольшой застой (кстати, напоминающий ситуацию перед тем, как появились методы нового поколения). Поэтому можно ожидать, что в ближайшие годы потребительская геномика будет по-прежнему полагаться на ДНК-микрочипы. Учитывая, то, насколько доступными они уже стали, даже введение новых революционных методов секвенирования вряд ли что-либо сильно поменяет на потребительском рынке. А значит, наступил момент, когда дело уже не в технологиях, с помощью которых получаются геномные данные, а в их интерпретации. Но это уже совсем другой разговор.
Код жизни: прочесть не значит понять
Код жизни: прочесть не значит понять
Секвенирование геномов — это не только самые современные приборы, но и большой объем вычислений на суперкомпьютерах!
Авторы
Редакторы
Последний год жизни авторы этой статьи посвятили созданию инфраструктуры по получению, хранению и анализу кода жизни — генетической информации, которая записана в молекуле ДНК. Что такое ДНК с точки зрения математика, каковы основные принципы построения компьютерной архитектуры для анализа огромных массивов генетической информации и что ждать в будущем от тотальной прозрачности и доступности теперь уже и нашего индивидуального кода жизни, — обо всём этом расскажет предлагаемая вашему вниманию статья.
Сегодня все знают, что информация, необходимая оплодотворённой яйцеклетке, чтобы развиться сначала в эмбрион, а потом и во взрослый организм, записана в молекулах ДНК, последовательность нуклеотидных остатков в которой можно представить в виде текста. В этом тексте, как и в любом другом, самое важное — это последовательность букв (в ДНК их, как известно, всего четыре). Совокупность всех молекул ДНК ядра клетки (каждая из которых, взаимодействуя с белками, образует отдельные хромосомы) называют ядерным генóмом (митохондрии, бывшие в незапамятные времена свободноживущими микроорганизамами, имеют свой собственный геном). К примеру, бактерии, обитающие у нас в кишечнике и помогающие переваривать пищу, имеют геном длиной порядка нескольких миллионов букв. Простая вошь — уже 500 миллионов, а геном человека составляет более трех миллиардов букв. Для сравнения, все четыре тома «Войны и мира» Толстого содержат около двух миллионов букв, — т. е., примерно эквивалентны генетической информации бактерии, а геном человека можно сравнить со всей библиотекой Толстого в Ясной Поляне. (С другой стороны, объём кинофильма в формате Blu-ray уже существенно превосходит размер генома, — так что говорить нужно, конечно, не только об объёме информации, но и о её «качестве».)
В первом приближении можно считать, что все клетки взрослого человека имеют один и тот же генетический текст. («Нормальное» исключение составляют половые клетки и лимфоциты, а патологическое — клетки раковой опухоли.) Так же, как и в книге, где фрагменты текста объединяются в главы, в геноме протяжённые последовательности нуклеотидов объединяются в гены, контролирующие функции клетки и организма [1]. Лишь десять лет назад геном человека был в общих чертах «прочитан» — в 2000 году две команды исследователей объявили о независимом друг от друга завершении проекта по секвенированию (определению последовательности ДНК) генома человека. Результат их работы сейчас считается «золотым стандартом» генома человека (то, с чем исследователи, как с эталоном, сравнивают вновь полученные генетические тексты). Общие затраты на эту работу оцениваются в интервале 3–10 миллиардов долларов [2].
Словарик
Читать — не перечитать
За прошедшие 10 лет технологии расшифровки генетических последовательностей развивались очень бурно, — в первую очередь, благодаря миниатюризации и автоматизации [3]. Следующее поколение методов секвенирования «информационных» молекул, будет, несомненно, связано с переходом к определению последовательности единичных молекул ДНК/РНК с применением нанотехнологических подходов. (Подробнее об этих технологиях, уже делающих первые уверенные шаги, «биомолекула» как-нибудь обязательно расскажет.)
Современная геномная лаборатория — например, Лаборатория геномики в Курчатовском Институте в Москве, — способна за один день «начитать» последовательность длиной до 20 миллиардов нуклеотидов. Что же касается стоимости таких работ, — прочтение одного человеческого генома уже подешевело почти до 10 000 долларов, — т. е., за 10 лет цена упала на 6 порядков (в миллион раз!). По оценкам экспертов, ценовой рубеж, когда персональная геномика войдёт в жизнь каждого из нас через медицину, страховки, работодателей и через прочие социальные институты, составляет 1000 долларов за индивидуальный геном. По всей видимости, он будет достигнут в ближайшие 5 лет.
Видимо, следует сделать оговорку — каждым из 6 миллиардов, достаточно обеспеченным, чтобы позволить себе тратить деньги на такие «пустяки». — Ред.
Внешние отличия, умственные способности и даже психологические особенности каждого человека в той или иной степени заложены в его геноме [4]. (Конечно, не стоит считать эти качества 100%-предопределёнными.) Считается, что основные генетические отличия одного человека от другого сосредоточены в однобуквенных заменах — своеобразных «опечатках» или «вариантах» текста ДНК, называемых однонуклеотидными полиморфизмами (ОНП). Как одна буква в названии книги «Война и мир» способна изменить его смысл (к примеру «Война и мор»), так и замены в генетических текстах могут привести к тому, что, например, одни люди будут болеть чаще, чем другие. Одни из нас — высокие и черноглазые, а другие — низкие и с голубыми глазами, и этим мы тоже обязаны генам. (И не только мы — например, «белая» окраска лошадей тоже обусловлена генетически [5].)
Зная генотип человека, теоретически можно предсказать многие его характерные черты, — не только цвет глаз и рост, но и предрасположенность к заболеваниям (именно это больше всего и интересует учёных и врачей) и даже к вредным привычкам [6]! Однако самое сложное здесь то, что большинство таких признаков определяется совокупностью большого, хотя и конечного числа «опечаток» в геноме, которые потребуется обнаружить. Результат появления той или иной «опечатки» не всегда бывает предсказуемым и понятным. Зачастую эффект от замены одной буквы слишком незначителен, и у исследователей нет ясности, как она в принципе может влиять на фенотип. Однако совокупность сотен и тысяч ОНП может эмпирически коррелировать с тем или иным признаком, хотя механизм, обусловливающий связь с признаком, может оставаться загадкой. Для этого необходимы данные о генотипах различных групп людей — как здоровых, так и больных, — чтобы иметь достаточную статистику для анализа отличий в геномах и поиска тех «опечаток», которые ответственны за склонность к заболеванию. (Подробнее о таких исследованиях, получивших название GWAS — genome-wide association studies — см. в статье «Загадочная генетика „загадочной болезни кожи“ — витилиго» [7].) Например, для проекта по исследованию рака почки генотипировано больше 12 тысяч человек. Таких проектов в мире можно насчитать уже несколько десятков, — то есть, всего насчитывается уже несколько сотен тысяч людей с установленными генотипами. Кстати говоря, уже и число полных геномов перевалило за тысячу [8].
Технология
Современные технологические платформы, предназначенные для чтения генетических текстов (для простоты такие приборы называют секвенаторами «нового поколения»), определяют последовательность не всей молекулы ДНК за раз, а лишь достаточно скромных её фрагментов — при большей длине, увы, возникает слишком много ошибок. Стадии секвенирования предшествует случайное «измельчение» ДНК на отрезки со средней длиной 500 нуклеотидов; само «считывание» осуществляется сразу с двух физических концов этого отрезка, в результате чего образуется пара фрагментов «текста» ДНК. Длина таких фрагментов, являющихся основным результатом работы секвенатора и на жаргоне именуемых «ридами» (от англ. read — читать), колеблется (в зависимости от производителя оборудования) в диапазоне 35–400 букв. Следует отметить, что большáя часть последовательности фрагмента неизбежно остаётся непрочитанной, поскольку длины «рида» (допустим, 100 букв) не хватает, чтобы перекрыть всю длину фрагмента (≈500 букв).
Подробнее об инструментальных основах секвенирования мы уже писали (см. «454-секвенирование (высокопроизводительное пиросеквенирование ДНК)» [3]); сейчас же разговор пойдёт главным образом о том, как скомбинировать из огромной библиотеки фрагментов текста «готовую» генетическую последовательность, как она записана в геноме. Современный прибор за один запуск длительностью около 10 дней способен сгенерировать до 300 миллионов пар «ридов» (рис. 1). Точная сборка этого «стройматериала» в геномную последовательность — проблема не из лёгких, и решаться она может двумя путями, в зависимости от наличия «эталонного» генома.

Рисунок 1. Секвенаторы SOLiD компании Applied Biosystems (Life Technologies) в лаборатории Queensland Centre for Medical Genomics, Австралия. Одна из миссий центра — «разрабатывать и улучшать технологию для рутинного анализа полной последовательности человеческого генома». Научных институтов, ориентированных на развитие «персонализованной медицины», с каждым годом становиться все больше и больше.
Задача первая: ресеквенирование
Возвращаясь к аналогии с произведением Льва Николаевича, ресеквенирование можно сравнить с поиском опечаток в исходном тексте «Войны и мира». Мы считаем, что геном изучаемого объекта имеет то же строение, что и «золотой стандарт» генома 2000-го года (хотя и этот эталон постоянно уточняется — сейчас доступна уже 37-я его версия). Сопоставляя участки текста, для каждого из многих миллионов «ридов» определяются его координаты на одной из хромосом: «страница», «абзац», «отступ слева» и т. д. В случае если обнаруживается расхождение с эталоном — опечатка, маленькая вставка или выпадение текста (на жаргоне такие отличия называют short indels — short insertions/deletions), — эти вариации включаются в отчёт о сравнении. Так, сравнивая миллионы и миллиарды «ридов» с исходным текстом, можно получить полный перечень отличий изучаемого генома от эталонного «золотого стандарта». Более того, если каждая буква исходного текста проверяется многократными прочтениями, это увеличивает статистическую достоверность найденных генетических особенностей и аномалий. Сегодня считается, что геном ресеквенирован с высоким «покрытием» (deep sequencing), если каждая его буква была прочитана в среднем 30 раз или более (30×).
С точки зрения биоинформатических алгоритмов, ресеквенирование — это относительно лёгкая процедура: для обработки данных от одного запуска прибора требуется всего около 10 часов работы программы на 20 процессорных ядрах и 20 Гб оперативной памяти.
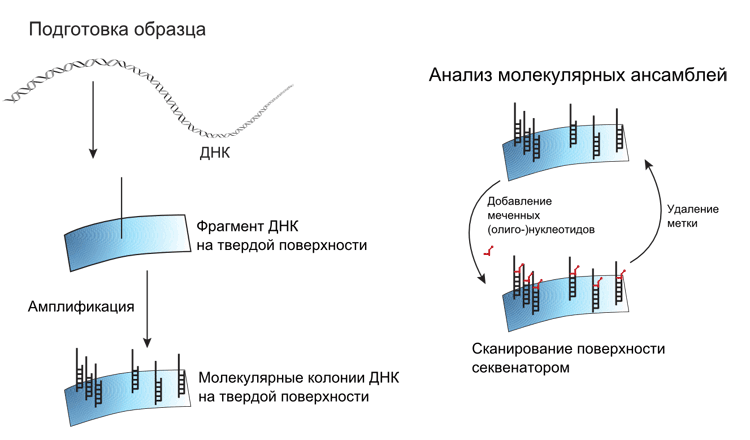
Рисунок 2. Общий принцип секвенирования нового («второго») поколения. Первый этап процесса подготовки образца состоит из фрагментации длинных молекул ДНК ультразвуком или каким-нибудь другим методом, не зависящим от последовательности ДНК, до размера 250–500 пар нуклеотидов. Далее следует стадия, во время которой концы ДНК-фрагментов приводятся в порядок: сначала удаляются или достраиваются выступающие концы молекул, затем лигируются адаптерные олигонуклеотиды. Полученная библиотека предварительно амплифицируется для увеличения представленности каждого фрагмента ДНК. Последний шаг — создание молекулярных колоний — клональная амплификация каждого фрагмента библиотеки на твердой поверхности. Все это происходит вне секвенатора. Расшифровка последовательности ДНК-фрагментов происходит через параллельный анализ миллионов и даже миллиардов молекулярных ансамблей в секвенаторе. В приборе стадии подачи реактивов для секвенирования (нуклеотидов или олигонуклеотидов с флуорофором и соответствующих ферментов) чередуются со стадиями сканирования поверхности и регистрации получаемых изображений.
Задача вторая: секвенирование de novo
Вторая задача является и экспериментально, и алгоритмически, и вычислительно более сложной — тут требуется реконструировать текст из набора «ридов», не имея эталона для сборки. Подход основан на том, что, в силу случайности разбиения молекул ДНК на фрагменты, при достаточно плотном «покрытии» обязательно найдутся несколько частично перекрывающихся «ридов», при совмещении которых текст будет постепенно наращиваться. «Подрастающий» текст (в данном случае называемый контигом) используется для поиска среди миллиарда «ридов» такого, который максимально (но не полностью) с ним перекрывается.
Процедура объединения контигов продолжается до тех пор, пока с обоих концов фрагмента генетического текста не начнутся области протяжённых повторов, характерные для «кончиков» хромосом. Если повтор имеет длину большую, чем длина «рида», то его длина, а значит, и точная последовательность, остаётся неизвестной. Однако здесь на помощь приходит информация о парности чтений: как правило, они находятся на более-менее известном расстоянии друг от друга. Таким образом, если одно из чтений пары попадает на один контиг, а второе — на другой, то эти контиги можно объединить в связку, называемую скаффолдом. Впоследствии непрочитанные «дыры» в скаффолдах можно будет прочесть другими методами. Сборка de novo является алгоритмически сложным и вычислительно затратным процессом.

Рисунок 3. Повторное секвенирование («ре-секвенирование») генома с целью выявления разнообразных структурных вариаций (однонуклеотидных полиморфизмов, или «снипов», а также инсерций, делеций, повторов, инверсий, транслокаций). В отличие от секвенирования неизвестных последовательностей de novo, при котором прочтения соотносятся друг с другом и собираются в контиги, для ре-секвенирования достаточно просто «картировать» прочтения на референсную последовательность, уже имеющуюся под рукой. Снипы выглядят как однонуклеотидные замены в коротких прочтениях, при этом количество прочтений с заменой говорит о состоянии аллеля — гомозиготном (все прочтения с заменой) или гетерозиготном (половина прочтений с заменой).
Такие задачи решаются с использованием теории графов, но идеального «сборщика» текстов для de novo-секвенирования ещё не создали. Основной проблемой сборки является наличие в генетических текстах длинных (от 200 до нескольких тысяч букв) элементов, содержащих повторы длиной от 4 до 150 нуклеотидных оснований. Очевидно, что именно из-за присутствия повторов текст во время сборки может оборваться. Для преодоления этого используют экспериментальные ухищрения, заключающиеся в генерации исходной библиотеки фрагментов со средней длиной не 500, а 3000 или даже 10000 букв. В этом случае существенно увеличивается вероятность захватить парой «ридов» уникальные участки текста, оставив повторы внутри.
Описав очень схематично схему алгоритма секвенирования, приступим к описанию вычислительных мощностей, которые авторы привлекают для анализа генетических текстов.
Секвенирование: без суперкомпьютеров не обошлось
«Сборка» текста генома из набора фрагментов, полученных на секвенаторе, — алгоритмически и вычислительно сложная задача, невозможная без использования суперкомпьютерных кластеров. Например, каждая сборка генома печёночного сосальщика, основанная на данных нескольких запусков секвенатора, требует до недели работы кластера из двух десятков узлов по 8 ядер и 8 Гб оперативной памяти в каждом (объединение по интерфейсу MPI). Однако одного запуска почти всегда недостаточно — таких сборок может быть несколько из-за необходимости подбора оптимальных параметров алгоритма и добавления новых экспериментальных данных. Есть и альтернативные варианты решения этой задачи, основанные не только на кластерах, но и популярных сегодня «облачных» вычислениях.
В целом, сборка de novo является более перспективным методом, чем ресеквенирование, и практически единственным подходом — когда эталонной последовательности генома исследуемого организма ещё не существует (как правило, для этого и проводится первое секвенирование). Оно позволяет выявлять существенные перестройки в геноме, обозревая его как одно целое. Впрочем, для многих практических целей и ресеквенирования бывает вполне достаточно.
Запрограммируй это
Читателю уже должно было стать ясно, что без серьёзных компьютерных мощностей решить задачу секвенирования генома шансов немного. Некоторые исследователи видят решение проблемы доступности дешёвых вычислений в так называемых «персональных суперкомпьютерах», под которыми имеются в виду системы на базе графических процессоров. Действительно, их специализация на операциях с векторами и массивная параллелизация находят всё более широкое применение во многих областях науки. В то же время относительно низкая цена и стоимость и владения такими компьютерами существенно снижают порог вхождения; их могут позволить себе не только институты, но и отдельные лаборатории.
Однако переход на новую технологию обязательно приносит с собой ряд проблем, и в биоинформатике они ощущаются особенно остро. В частности, часто необходимо использовать специальные алгоритмы, требующие от программиста знания архитектуры графических процессоров; в то же время большинство программного обеспечения в биологии разрабатывается биологами, для которых программирование — лишь дополнительный навык. Это же обусловливает и приверженность биологов к скриптовым языкам программирования: часто требуется написать простую программу «на раз» — только для проверки очередной гипотезы. Традиционно используемый в биоинформатике язык Perl на настоящий момент не имеет доступа к OpenCL (программный комплекс для облегчения программирования графических процессоров), хотя некоторые другие языки, например, Python или Java, уже оснащены привязкой к этому фреймворку.
Можно назвать три основных особенности использования графических сопроцессоров в геномной биоинформатике. Во-первых, это текстовый формат геномных данных, в то время как единого стандарта для их представления в удобном (с точки зрения вычислений, компактном и быстром) цифровом виде до сих пор нет. Во-вторых, высокая «квантуемость» данных («ридов» секвенатора, полиморфизмов и др.) способствует многопоточной обработке. И, в-третьих, огромные требования к оперативной памяти, запас которой непосредственно на графическом ускорителе пока относительно мал; это может привести к дополнительному усложнению и без того порой неочевидных алгоритмов.
Требования к аппаратному обеспечению, накладываемые практическими задачами, почти всегда опережают реальные возможности вычислительных машин. Для многих приложений биоинформатики — таких как сборка геномов de novo — часто не хватает ресурсов даже самых современных кластеров, включающих сотни вычислительных узлов, соединенных быстрой сетью Infiniband.
Возможное решение проблемы компьютерных мощностей — глобальная грид-инфраструктура, объединяющая десятки суперкомпьютерных центров и позволяющая использовать их мощности через единый интерфейс. Кроме того, грид-технология позволяет создавать распределённое хранилище данных, — а ведь когда речь идёт о геномике, дискового пространства не бывает много. Последнее время это направление распределённых вычислений очень активно развивается, — например европейский проект EGEE является примером создания крупнейшей грид-инфраструктуры, объединяющей участников из более чем 50 стран и включающей в себя более 260 компьютерных центров. Общее количество вычислительных ядер в этой сети более 150 тысяч, а дисковое пространство превышает 28 петабайт. Возможно, использование грид-технологий сможет отодвинуть границу доступных задач в биоинформатике уже в самом ближайшем будущем.
Секвенирование геномов в России

Надо сказать, что в нашей стране дела с полногеномным секвенированием не так уж и плохи. Первые секвенаторы «нового поколения» — приборы Applied Biosystems (сейчас в составе Life Technologies) SOLiD и Illumina GAII — заработали в Российском научном центре «Курчатовский институт» в начале 2009 года. В Лаборатории геномного анализа, которую возглавляет Егор Прохорчук (специально для этого вернувшийся из-за границы), и был расшифрован первый российский геном человека. Разумеется, русского человека. Предварительные результаты этой работы были опубликованы в отечественном журнале Acta Naturae [10]. В настоящее время в лаборатории ведется работа по расшифровке ещё нескольких человеческих геномов в рамках крупного международного проекта по исследованию раковых заболеваний.
Сейчас Курчатовский институт (со своим отдельным финансированием и руководством, не зависящим от Российской академии наук) является самым крупным российским геномным центром, заметным даже по европейским меркам (но не по американским или китайским, где число секвенаторов только одного типа может переваливать за сотню). Но и другие институты, уже в составе РАН, не отстают от «Курчатника» (см. таблицу). Как известно, Россия прирастает Сибирью. Сибиряки из Института химической биологии и фундаментальной медицины, что находится в новосибирском Академгородке, имеют в своём распоряжении две, в некотором смысле комплементарные, платформы для высокопроизводительного секвенирования — SOLiD и 454 FLX Titanium («Roche — всем хорош»). В институте работает центр коллективного пользования «Секвенирование ДНК», которым руководит доктор Игорь Морозов. Именно в этом ЦКП и осуществляется несколько различных научных проектов с использованием высокопроизводительных секвенаторов.
Медицинские НИИ тоже участвуют в полногеномных исследованиях. В НИИ физико-химической медицины в лаборатории профессора Вадима Говоруна прибор Applied Biosystems SOLiD 4 будет работать на благо российской биомедицины.
Также пара мощных приборов двух конкурирующих компаний (Life Technologies и Illumina) устанавливаются в Москве в Институте общей генетики РАН им. Н. И. Вавилова в лаборатории профессора Евгения Рогаева, известного и за рубежом российского ученого, прославившегося своими исследованиями болезни Альцгеймера, а также расшифровкой ДНК из останков царской семьи [11].
Ещё несколько приборов разбросанно по московским и периферийным институтам; один SOLiD скоро заработает в коммерческой компании, специализирующейся на предоставлении услуг по высокопроизводительному секвенированию и генотипированию. Такая картина не может не вселять определенный оптимизм в стороннего наблюдателя, следящего за вялым возрождением отечественной науки. Остается только с нетерпением ждать появления публикаций в престижных журналах, ведь у работ с привлечением (пока еще!) передовых методов и оборудования шансов на успех должно быть больше.
Заключение
Как же будет выглядеть персональная геномика через несколько лет? Нам видится, что буквально в каждом крупном городе будет лаборатория по чтению индивидуальных геномов. Скорее всего, не стоит опасаться того, что индивидуальная информация станет доступна лаборанту, обслуживающему секвенатор — он все равно не сможет её интерпретировать. Хотя, безусловно, сохранности персональной тайны пациента следует уделить самое пристальное внимание. Последовательности геномов будут поступать в единый информационный центр, который проведёт автоматизированный анализ и сравнение с геномами других людей, для которых уже создана история болезни. На основании этого можно будет делать индивидуальный прогноз для каждого пациента, который, быть может, ещё и не пациент вовсе, а всего лишь новорожденный.
Более того, все болезни, физические параметры, психофизиологические особенности, наблюдаемые у него в будущем, также можно будет занести в его персональный файл и соотнести с генетическим текстом. Приходя на приём к врачу, человек получит рекомендации по лечению и приёму лекарств, наиболее соответствующих его метаболизму и предрасположенностям.
| Название учреждения | Платформа |
|---|---|
| РНЦ «Курчатовский институт», Москва | Illumina GA IIx (×3) Applied Biosystems SOLiD 4 (×2) |
| НИИ химической биологии и фундаментальной медицины СО РАН, Новосибирск | Applied Biosystems SOLiD 4 454/Roche FLX Titanium |
| Инновационно-технологический центр «Биологически активные соединения и их применение» РАН, Москва | llumina GA IIx |
| Институт общей генетики им Н. И. Вавилова, Москва | Applied Biosystems SOLiD 4 llumina HiSeq 2000 |
| Институт молекулярной биологии им. В. А. Энгельгардта РАН, Москва | llumina GA IIx |
| Генаналитика, Москва | Applied Biosystems SOLiD 4 |
| Секретная станция переливания крови, Киров | 454/Roche FLX Titanium |
| Лимнологический институт СО РАН, Иркутск | 454/Roche FLX Titanium |
| Центр «Биоинженерия» РАН, Москва | 454/Roche FLX Titanium |
| НИИ физико-химической медицины, Москва | Applied Biosystems SOLiD 4 |
| Биологический факультет МГУ им. М. В. Ломоносова, Москва | Applied Biosystems SOLiD 4 |
. Над всем этим неумолимо нависает призрак киберпанка, предвосхищая растворение человека в океане цифр и знаков. Большой брат будет следить и за тобой. Но только если ты этого захочешь. Это наступит неотвратимо, но не через один год жизни...
Первоначально сокращённый вариант этой статьи был опубликован в журнале «Суперкомпьютеры» [9].
Статья написана в соавторстве с Чекановым Н.Н. и Теслюком А.Б. при участии коллектива «биомолекулы». Врезку, таблицу, картинки и «словарик» подготовил Павел Натальин.


