Вторичные тромботические микроангиопатии
ФГБОУ ВО «Первый Санкт-Петербургский государственный медицинский университет им. акад. И.П. Павлова» МЗ РФ, кафедра нефрологии и диализа, Санкт-Петербург, Россия
Тромботическая микроангиопатия (ТМА) – клинико-морфологический синдром, относящийся к спектру заболеваний с эндотелиальным повреждением в качестве этиологического фактора. В статье рассматриваются вопросы классификации, особенности и патогенез вторичных ТМА, стратегии современной диагностики и лечения. Клиническая картина вторичных ТМА не имеет специфических черт. Условием постановки клинического диагноза ТМА является наличие как минимум двух симптомов: тромбоцитопении и микроангиопатической гемолитической анемии. Развитие вторичных ТМА связано с широким спектром различных заболеваний и состояний, рассмотренных в данном обзоре. Наиболее важно вовремя распознать их, т.к. это значительно улучшает прогноз. Своевременная диагностика и лечение ТМА, ассоциированной с беременностью, показывают хорошие результаты. С другой стороны, прогноз при ТМА, ассоциированной с трансплантацией гемопоэтических стволовых клеток, и ТМА, ассоциированной с химиотерапией, остается неудовлетворительным. В настоящее время не существует общепринятых стандартов лечения вторичных ТМА.
Введение
Тромботическая микроангиопатия (ТМА) – этиологически гетерогенный клинико-морфологический синдром, характеризующийся поражением сосудов микроциркуляторного русла.
Гистологически ТМА – это особый тип повреждения микрососудов, представленный отеком эндотелиальных клеток с их отслойкой от базальной мембраны, расширением субэндотелиального пространства с накоплением в нем аморфного мембраноподобного материала с образованием тромбов, содержащих тромбоциты и фибрин, что приводит к окклюзии просвета сосуда, вызывая развитие ишемии органов и тканей [1].
ТМА проявляется тромбоцитопенией, возникающей вследствие усиленного потребления тромбоцитов при образовании множественных микротромбов, микроангиопатической гемолитической Кумбс-негативной анемией (механический гемолиз), лихорадкой и в зависимости от вовлечения того или иного участка сосудистого русла – почечной недостаточностью, кардиальными осложнениями, дыхательной недостаточностью, нарушениями зрения, панкреатитом, ишемией кишечника [1]. До появления эффективных методов лечения смертность была высокой, достигая 72–94%.
ТМА классифицируют на первичные и вторичные. Первичные ТМА включают тромботическую тромбоцитопеническую пурпуру, типичный и атипичный гемолитико-уремический синдром (ГУС).
Первичные ТМА
Тромботическая тромбоцитопеническая пурпура (ТТП)
В основе патогенеза ТТП лежит дефицит металлопротеиназы ADAMTS13 (а disintegrin-like and metalloprotease with thrombospondin type I motif 13), которая в норме расщепляет образуемые эндотелием крупные мультимеры фактора Виллебранда (VWF – von Willebrand factor). Дефицит этого фермента приводит к циркуляции в плазме крови пациентов сверхкрупных мультимеров VWF с образованием в системе микроциркуляции тромбов. Дефицит ADAMTS13 может быть обусловлен мутациями в гене ADAMTS13 при редких врожденных формах ТТП (синдром Upshaw–Schulman) или появлением в циркуляции аутоантител к ADAMTS13, являющихся его ингибиторами, что имеет место при более часто встречаемых приобретенных формах ТТП.
Отличительные особенности ТТП:
Главная составляющая лечения ТТП – сеансы истощающего плазмообмена. При выявлении ингибиторных антител к ADAMTS13 – добавление к терапии глюкокортикоидов. При неэффективности проводимого лечения может быть использована иммуносупрессивная терапия ритуксимабом (off-label) [3].
Типичный гемолитико-уремический синдром (ГУС)
Типичная (пост-диарейный, STEC – Shiga Toxin-Producing Escherichia Coli) ТМА, опосредованная повреждающим эндотелий действием шига-токсина кишечной палочки (STEC – шига-токсин продуцирующий штамм E. сoli, обычно O157:H7 или O104:H4 серотипов), с преимущественным вовлечением почек – развитием острого повреждения почек (ОПП).
Скрининг на STEC-ГУС необходим всем больным с признаками поражения желудочно-кишечного тракта, особенно с диареей. Лабораторные исследования следует выполнять в первые сутки госпитализации больного до начала антибактериальной терапии. Для диагностики STEC-ГУС показаны посев кала для выявления культуры STEC, определение шига-токсина в кале и сыворотке крови. В отношении лечения рекомендуются адекватная антибактериальная терапия (предпочтительны бактериостатические антибиотики), коррекция водно-электролитных нарушений и при необходимости своевременное начало диализных методов лечения [1].
Атипичный гемолитико-уремический синдром (аГУС)
аГУС – заболевание, опосредованное дисфункцией системы регуляции комплемента с неконтролируемой активацией его альтернативного пути. аГУС чаще всего имеет в основе генные мутации белков – регуляторов системы комплемента: CFH (фактор H) у 20–25% пациентов, MCP (мембранный кофакторный протеин) – у ≈15% и CFI (фактор I) – у ≈10%. Мутации фактора В (CFB) встречаются крайне редко (1%), в то время как мутации C3 фракции комплемента встречаются среди 10% пациентов. Редкими являются мутации гена тромбомодулина (THBD). Терапия включает сеансы плазмообмена и введение экулизумаба (гуманизированного моноклонального антитела к C5 фракции терминальной стадии каскада комплемента).
Диагноз аГУС – это диагноз исключения. Он устанавливается на основании характерной клинической картины и должен быть подтвержден лабораторными данными, исключающими другие причины ТМА.
 В данном обзоре мы более подробно остановимся на вторичных ТМА.
В данном обзоре мы более подробно остановимся на вторичных ТМА.
Вторичные ТМА
ТМА определяют как вторичные, когда их развитие ассоциировано с различными заболеваниями или состояниями. Наиболее частые причины вторичных ТМА – беременность, аутоиммунные заболевания, злокачественные новообразования, прием некоторых лекарственных препаратов, инфекции, трансплантация сóлидных органов или костного мозга.
Тромботическая микроангиопатия в акушерстве
Введение
Преэклампсия и HELLP-синдром являются грозным осложнением беременности. В настоящее время они рассматриваются как варианты тромботической микроангиопатии (ТМА). Наиболее грозным представителем ТМА является атипичный гемолитико-уремический синдром (аГУС), к развитию которого предрасполагают генетические аномалии в системе иммунитета. Установлено, что беременность сама по себе может активировать патологический иммунный ответ, причем выраженность активации возрастает при наличии акушерских осложнений, достигая максимума у пациенток с преэклампсией. Генетический дефект в сочетании с преэклампсией приводит к неконтролируемой активации иммунного ответа, являющегося при акушерском аГУС основой развития полиорганной недостаточности, которая не может быть устранена без специфического лечения.
Критерии диагноза ТМА
ТМА представляет собой синдром, в основе которого лежит повреждение эндотелия сосудов микроциркуляторного русла (МЦР) различными способами, но имеющий сходные проявления и диагностические признаки. Результатом эндотелиального повреждения служит тромботическая микроангиопатия — особый тип поражения мелких сосудов, представленный их тромбозом и воспалением сосудистой стенки.
Морфологическая картина ТМА: отек эндотелиальных клеток, их отслойка от базальной мембраны (эндотелиоз), некроз, деструкция, расширение субэндотелиального пространства, тромбы в просвете капилляров и артериол, содержащие тромбоциты и фибрин, нередко с полной окклюзией просвета сосудов.
Клинико-лабораторные признаки ТМА:
Тромботические микроангиопатии классифицируют на первичные и вторичные.
Вторичные ТМА вследствие следующих состояний:
Критерии диагноза аГУС в акушерстве
Диагноз аГУС в акушерстве — это диагноз исключения. Дифференциальная диагностика с другими формами ТМА приведена в табл. 1.
Положительный результат при бактериологическом исследовании кала: посев на среду для выявления STEC (Mac Conkey для 0157:Н7), определение в образцах фекалий ДНК энтеро-геморрагических E.coli методом ПЦР; выявление в сыворотке антител к липополисахаридам наиболее распространенных в данном регионе серотипов E.coli.
Наследственная или приобретенная ТТП
Дефицит ADAMTS-13 — активность менее 10%, антитела к ADAMTS-13
Беременность. Исключить преэклампсию и HELLP-синдром
Ферменты печени, срок гестации, критерии преэклампсии и тяжелой преэклампсии, положительная динамика непосредственно после родоразрешения
Аутоиммунные заболевания (системная красная волчанка, антифосфолипидный синдром)
Анти-ДНК-антитела, антинуклеарные антитела, антитела к кардиолипину IgG и/или IgM изотипов, антитела к (32 GP 1 IgG и/или IgM изотипов с помощью стандартизованного иммуноферментного метода, волчаночный антикоагулянт стандартизованным коагулологическим методом
Положительные результаты иммунного блоттинга на ВИЧ- инфекцию
Наличие очага инфекции и полиорганной недостаточности (острое изменение по шкале SOFA >2 баллов)|
Генетическое исследование и биопсия почки не являются необходимыми для установления диагноза аГУС и не играют роли для решения вопроса о тактике лечения больного.
Преэклампсия и HELLP-синдром являются специфическими ассоциированными с беременностью формами ТМА. Всем пациенткам, госпитализированным с диагнозом тяжелая преэклампсия и/или HELLP-синдром, необходимо до родоразрешения исследовать ЛДГ, гаптоглобин в сыворотке крови и шизоциты в мазке периферической крови, а также определить количество тромбоцитов и уровень креатинина.
Истинные тяжелая преэклампсия и HELLP-синдром требуют родоразрешения с целью элиминации секретирующегося анти-ангиогенного фактора sFlt-1 плаценты.
Поскольку термин «HELLP-синдром» — собирательное понятие и его причины до конца не выяснены, тактика родоразрешения и интенсивной терапии строится в соответствии с тактикой при тяжелой преэклампсии (родоразрешение). В этом случае диагноз формулируется в соответствии с МКБ Х — «Тромботическая микроангиопатия (HELLP-синдром)».
Принципы и схемы терапии
При развитии клиники HELLP-синдрома в послеродовом периоде необходимо строить тактику интенсивной терапии в зависимости от следующих клинических вариантов:
Вариант 1. У пациентки сохранены: сознание, диурез более 0,5 мл/кг/ч (вне зависимости от цвета мочи), стабильная гемодинамика (или с тенденцией к артериальной гипертензии), отсутствует геморрагический синдром любой локализации. При лабораторном исследовании выявлены тромбоцитопения, повышены уровни АСТ, АЛТ, ЛДГ, массивного внутрисосудистого гемолиза нет. Плазменные факторы свертывания в норме. В данном случае, в течение суток оценивается динамика клинико-лабораторных
проявлений HELLP-синдрома и при отсутствии отрицательных проявлений интенсивная терапия ограничивается базовой терапией преэклампсии и инфузией кристаллоидов Пациентка получает нутритивную поддержку и активизируется. Проводится тромбопрофилактика НМГ при количестве тромбоцитов более 70000 в мкл.
Вариант 2. Уже с первых часов после родоразрешения развивается клиника острой печеночной недостаточности (тромбоцитопения, рост АСТ, АЛТ, коагулопатия, кровотечение, шок, ОПН, ОРДС и т.д.), в основе, которой лежит некроз печени (подкапсульная гематома). Требует проведения комплексной посиндромной интенсивной терапии острой печеночной недостаточности в условиях многопрофильного ЛПУ с возможностью хирургического лечения.
Вариант 3. Развитие массивного внутрисосудистого гемолиза (свободный гемоглобин в крови и моче, анемия) уже в первые часы осложняется развитием ОПН (по шкалам RIFLE, AKIN, KDIGO) и требует проведения заместительной почечной терапии. Противопоказано применение магния сульфата и инфузионной терапии. Требует проведения комплексной посиндромной интенсивной терапии ОПН в условиях многопрофильного ЛПУ. При сохранении или прогрессировании симптомов ТМА (тромбоцитопения и МАГА) в течение 48 часов следует, как один из вероятных диагнозов рассматривать аГУС и проводить соответствующую терапию.
Вариант 4. В исключительных случаях верификации диагноза ТТП в послеродовом периоде на основании сочетания признаков HELLP-синдрома, нарастающей тромбоцитопении, симптомов поражения почек и/или ЦНС со снижением активности ADAMTS-13 менее 10% показана инфузия свежезамороженной плазмы и проведение плазмообмена«.
Вариант 5. Женщинам, перенесшим акушерскую ТМА (преэклампсия, HELLP-синдром), следует устанавливать диагноз аГУС, если после родоразрешения их состояние не улучшается или ухудшается, в короткие сроки приводя к формированию
полиорганной недостаточности, что свидетельствует о персистировании ТМА с генерализацией микроангиопатического процесса.
В первую очередь о возможном аГУС следует думать при развитии тяжелого HELLP-синдрома с признаками внепеченочного поражения, особенно если родоразрешение не сопровождается положительной динамикой состояния пациентки, несмотря на лечение в соответствии с клиническими рекомендациями (протоколом лечения) МЗ РФ «Гипертензивные расстройства во время беременности, в родах и послеродовом периоде. Преэклампсия. Эклампсия» 2016 г. Быстрое нарастание анемии при отсутствии выраженной кровопотери свидетельствует об усилении микроангиопатического гемолиза, что, как правило, сопровождается усугублением тромбоцитопении и стремительным ухудшением функции почек, приводящим к развитию олигурической.
Родильницам с установленным диагнозом аГУС следует назначать патогенетическую терапию, направленную на блокирование С5-компонента системы комплемента, играющего ключевую роль в развитии данного осложнения. Антикомплементарная терапия проводится согласно рекомендациям по лечению аГУС взрослых.
Экулизумаб — рекомбинантное гуманизированное моноклональное антитело класса Ig G к С5 компоненту комплемента. Препарат блокирует расщепление С5, препятствуя образованию мембрано-атакующего комплекса и предотвращая тем самым повреждение эндотелия и, следовательно, прекращая процессы микроциркуляторного тромбообразования. Применение Экулизумаба приводит к обратному развитию ТМА и/или предупреждает прогрессирование поражения почек. Критериями эффективности терапии Экулизумабом являются прекращение микроангиопатического гемолиза (снижение уровня ЛДГ до нормальных значений) и нормализация числа тромбоцитов, а также улучшение функции почек.
Начальный курс терапии Экулизумабом рассчитан на 5 недель, далее подразумевается переход на цикл поддерживающего лечения.
Индукционный курс: 1 раз в неделю вводят по 900 мг Экулизумаба на протяжении недель. На пятой неделе дозу увеличивают до 1200 мг.
Поддерживающий этап: каждые 14 (плюс/минус 2 дня) дней вводят по 1200 мг.
В ожидании Экулизумаба родильницы с установленным диагнозом аГУС должны получать в случае необходимости почечную заместительную терапию при наличии ОПН. Свежезамороженная плазма в больших объемах у пациенток с тяжелой преэклампсией может вызвать перегрузку объемом (TACO-синдром) или развитие иммунного TRALI-синдрома и в отсутствие клинических проявлений коаулопатии и кровотечения противопоказана! Свежезамороженная плазма применяется только при верификации диагноза ТТП.
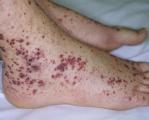
Болезнь Мошковица – это полисиндромная патология, сочетающая в себе тромбоцитопению, гемолитическую анемию, окклюзию артериол с развитием ишемических поражений органов. Клинически протекает с лихорадкой, кровотечениями, неврологическими нарушениями, абдоминальным синдромом. Возможно развитие ТИА, инфаркта миокарда и почек, гепатита, мезентериальной ишемии. Диагностика основана на исследовании крови (ОАК, тромбоциты, ЛДГ, активность металлопротеиназы), биоптатов почек, кожи, костного мозга. Лечение предполагает использование дезагрегантов, антикоагулянтов, глюкокортикоидов; проведение плазмафереза, переливание плазмы, по показаниям – гемодиализ.
МКБ-10
Общие сведения
Причины
Этиология болезни Мошковица остается предметом исследования в гематологии. Важным прорывом последних десятилетий в этом вопросе служит выявление связи ТТП с недостаточностью фермента ADAMTS-13 – металлопротеазы, расщепляющей фактор Виллебранда. Нарушение ферментной активности может быть вызвано как врожденными, так и приобретенными причинами:
Патогенез
Дефицит или ингибирование ADAMTS-13 приводит к повышенной активности VWF (фактора фон Виллебранда), который начинает стимулировать неконтролируемую внутрисосудистую агрегацию тромбоцитов и тромбообразование в микроциркуляторном русле. Количество циркулирующих тромбоцитов уменьшается, развивается тромбоцитопения.
Одновременно происходит пролиферация эндотелия мелких сосудов, что вызывает дополнительное сужение их просвета. Механическое затруднение для тока крови и высвобождение различных эндотелиальных факторов способствует фрагментации эритроцитов с развитием внутрисосудистого гемолиза.
Агрегаты тромбоцитов вызывают закупорку артериол и капилляров головного мозга, сердца, почек, легких. На коже и слизистых возникает геморрагическая пурпура, внутренние органы увеличиваются, в них обнаруживаются отложения гемосидерина. На фоне микроангиопатии формируются ишемические и некротические изменения тканей, возникают инфаркты сердечной мышцы, церебральной ткани, почечной паренхимы.
Классификация
С точки зрения влияния этиологических факторов, болезнь Мошковица подразделяется на наследственные и приобретенные варианты. Эти группы (особенно приобретенная форма) крайне неоднородны, отличаются по времени дебюта, доброкачественности течения, прогнозу:
1. Наследственная. Представлена синдромом Апшоу–Шульмана с аутосомно-рецессивным механизмом наследственной передачи. Чаще манифестирует в детском возрасте, однако известны эпизоды начала заболевания в 35 лет. Течение хроническое, рецидивирующее.
2. Приобретенные формы ТТП.
Симптомы болезни Мошковица
В большинстве случаев заболевание манифестирует остро и внезапно. Часто отмечается простудоподобное продромальное состояние. Клиника развивается стремительно. Классическая форма болезни Мошковица характеризуется пентадой признаков: тяжелой тробоцитопенией, гемолитической анемией, неврологическими нарушениями, лихорадкой и почечным синдромом.
В дебюте ТТП у 35% больных возникает абдоминальный синдром: рвота, выраженные боли в животе, свидетельствующие о мезентериальной ишемии. Типична лихорадка. Может выявляться гепатит, панкреатит, желтуха, спленомегалия. Иногда заболевание манифестирует с инфаркта миокарда, ОРДС, рабдомиолиза, гангрены.
Тромбоцитопения сопровождается появлением петехиальной сыпи на коже, кровотечений различной степени выраженности и локализации (десневые, носовые, меноррагии, мелена, кровохарканье, кровоизлияние в подпаутинное пространство). Церебральные и почечные нарушения отражают ишемические поражения органов. В неврологическом статусе отмечаются цефалгии, судороги, нарушения сознания. Могут возникать парезы и гемиплегия, атаксия, афатические расстройства. В тяжелых случаях возможна кома.
Признаками почечного синдрома служат массивные отеки, артериальная гипертензия, гематурия. Нарастание олигурии, электролитного дисбаланса и азотемии свидетельствует о развитии острой почечной недостаточности. Чаще болезнь Мошковица протекает остро или подостро. Возможно рецидивирующее течение.
Осложнения
Без экстренного назначения адекватного лечения смертность от болезни Мошковица составляет 85-100%. Летальные исходы обусловлены ишемическим поражением органов-мишеней (острый инфаркт миокарда, ОНМК, инфаркт почек), геморрагическим синдромом (церебральные кровоизлияния, внутренние кровотечения), органной дисфункций (ОПН, СН). Описаны случаи внезапной сердечной смерти, связанные с фатальными аритмиями, кардиогенным шоком. В поздних стадиях развивается ДВС-синдром.
Диагностика
Сложность распознавания болезни Мошковица объясняется отсутствием патогномоничной клинической картины. Больные часто поступают в стационар с клиникой инсульта, острой коронарной недостаточности, абдоминальной ишемии и безуспешно лечатся от соответствующих синдромов. Между тем, наряду с осмотром врача-невролога, кардиолога, нефролога, пациентам необходима консультация гематолога:
Дифференциальный диагноз
На основании лабораторно-инструментальных данных дифференциальная диагностика болезни Мошковица осуществляется с:
Лечение болезни Мошковица
При малейшем подозрении на ТТП рекомендуется незамедлительно начинать патогенетическое лечение. Пациенты, как правило, госпитализируются в ОРИТ, где проводится постоянный мониторинг клинико-лабораторных показателей. Терапия болезни Мошковица включает несколько направлений:
При рефрактерных к стандартной терапевтической схеме формах болезни Мошковица применяются цитостатики, иммунодепрессанты, моноклональные антитела. В целях снижения риска рецидивов показана спленэктомия.
Прогноз и профилактика
Болезнь Мошковица ‒ тяжелая, сложно диагностируемая патология с агрессивным течением. Без своевременного лечения летальность очень высока. При быстрой постановке диагноза и раннем начале лечения (проведении экстракорпоральной гемокоррекции) удается спасти свыше 80% больных. В дальнейшем для пролонгирования ремиссии необходима постоянная поддерживающая терапия. Часто сохраняются остаточные изменения в виде энцефалопатии, ХПН. Профилактика ТТП не разработана. При наследственных формах проводится генетическое консультирование.
Тромботические микроангиопатии
Тромботическая микроангиопатия – клинический синдром, для которого характерны:
Морфологически тромботическая микроангиопатия (ТМА) определяется как уплотнение сосудистой стенки с набуханием или отделением эндотелиальных клеток от базальной мембраны и отложением гиалиновых депозитов в субэндотелиальном пространстве, внутрисосудистые тромбоцитарные тромбы и окклюзия сосудов [1]. Повреждение эндотелия сосудов при ТМА индуцирует процесс образования внутрисосудистых тромбоцитарных тромбов мелких сосудов. Потребление тромбоцитов приводит к развитию тромбоцитопении, сужение просвета сосудов вызывает микроангиопатическую гемолитическую анемию (происходит механическое разрушение эритроцитов), ишемию важнейших органов.
Выделяют также ТМА-ассоциированные синдромы:
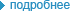
ТМА может развиваться:
Обязательными элементами заболеваний, относящихся к ТМА и ТМА-ассоциированным синдромам, являются микроангиопатическая гемолитическая анемия и тромбоцитопения.
МикроАнгиопатическая Гемолитическая Анемия (МАГА) характеризуется:
Шистоцитоз (превышение нормального уровня количества шистоцитов) – необходимый диагностический признак тромботической микроангиопатии.
Шистоциты – это фрагменты эритроцитов, выявляемые в мазке крови, в виде:
Международный Совет по Стандартизации в Гематологии (ICSH) разработал рекомендации по идентификации шистоцитов. Было предложено также считать шистоцитами микросфероциты (при наличии в мазке крови шистоцитов другой формы) [2].
Диагностические критерии тромботической тромбоцитопенической пурпуры (диагностическая диада) при отсутствии другой выявленной причины:
Диагностические критерии гемолитико-уремического синдрома (диагностическая триада):
Диагностические критерии HELLP-синдрома:
ЗНАЧЕНИЕ ТРОМБОТИЧЕСКОЙ МИКРОАНГИОПАТИИ В ПАТОГЕНЕЗЕ АКУШЕРСКИХ ОСЛОЖНЕНИЙ
Акушерство, гинекология и репродукция. 2015; N2: c.62-71
Тромботическая микроангиопатия представляет собой одно из наиболее тяжело протекающих тромботических осложнений, характеризующееся поражением микрососудов различных органов и сопровождающееся тромбоцитопенией и гемолитической анемией. Термин тромботическая микроангиопатия вобрал в себя несколько нозологий, для которых характерны разные механизмы возникновения микротромбоза. В настоящее время к тромботической микроангиопатии относят тромботическую тромбоцитопеническую пурпуру (ТТП), гемолитико-уремический синдром (ГУС), гепарин-индуцированную тромбоцитопению, HELLP-синдром. Одним из важнейших триггеров к возникновению тромботической микроангиопатии является беременность. Этот факт открывает широкие перспективы к изучению патогенеза тромботической микроангиопатии в контексте физиологических изменений гемостаза во время беременности. В то же время открытие молекулярных механизмов тромботической микроангиопатии позволяет по-новому взглянуть на патогенез тромботических осложнений, связанных с беременностью, а также на патогенез так называемых плацентарных акушерских осложнений, в том числе тяжелых форм преэклампсии, преждевременной отслойки нормально расположенной плаценты, септического шока.
Статья поступила: 23.04.15 г.; в доработанном виде: 25.05.2015 г.; принята к печати: 22.06.2015 г.
Конфликт интересов
Авторы заявляют об отсутствии необходимости раскрытия финансовой поддержки или конфликта интересов в отношении данной публикации.
Все авторы сделали эквивалентный вклад в подготовку публикации.
Для цитирования
Акиньшина С.В., Бицадзе В.О., Гадаева З.К., Макацария А.Д. Значение тромботической микроангиопатии в патогенезе акушерских осложнений. Акушерство, гинекология и репродукция. 2015; 2: 62-71.
THROMBOTIC MICROANGIOPATHY IN THE PATHOGENESIS OF OBSTETRIC COMPLICATIONS
Akinshina S.V., Bitsadze V.O., Gadaeva Z.K., Makatsariya A.D.
First Moscow State Medical Sechenov University of the Ministry of Health Russian Federation
Summary
Thrombotic microangiopathy is one of the most serious thrombotic complications characterized by microvascular thrombosis in various organs and accompanied by thrombocytopenia and hemolytic anemia. The term thrombotic microangiopathy has incorporated several nosology, which are characterized by different mechanisms of microvascular thrombosis. Currently thrombotic microangiopathy include thrombotic thrombocytopenic purpura (TTP), hemolytic uremic syndrome (HUS), heparin-induced thrombocytopenia, HELLP-syndrome. Pregnancy presents one of the key triggers to the development of thrombotic microangiopathy. This fact gives us a significant opportunity to study the pathogenesis of thrombotic microangiopathy in the context of the physiological changes of hemostasis during pregnancy. At the same time the discovery of molecular mechanisms of thrombotic microangiopathy allows for a new research on the field of pathogenesis of thrombotic complications associated with pregnancy, as well as the pathogenesis of so-called placental obstetric complications, including severe preeclampsia, premature detachment of normally situated placenta, septic shock.
Received: 23.04.15; in the revised form: 25.05.2015; accepted: 22.06.2015.
Conflict of interests
The authors declared that they do not have anything to disclosure regarding funding or conflict of interests with respect to this manuscript.
All authors contributed equally to this article.
For citation
Akinshina S.V., Bitsadze V.O., Gadaeva Z.K., Makatsariya A.D. Thrombotic microangiopathy in the pathogenesis of obstetric complications. Akusherstvo, ginekologiya i reproduktsiya / Obstetrics, gynecology and reproduction. 2015; 2: 62-71 (in Russian).
Corresponding author
Address: ul. Zemlyanoi Val, 62-1, Moscow, Russia, 109004.
E-mail address: svetlana_akin@mail.ru (Akinshina S.V.).
Ключевые слова: Тромботическая микроангиопатия, преэклампсия, HELLP синдром, материнская смертность.
ГБОУ ВПО «Первый МГМУ имени И.М. Сеченова» Минздрава России, Москва
Понятие о тромботической микроангиопатии и тромботической тромбоцитопенической пурпуре
Тромботическая тромбоцитопеническая пурпура (ТТП) в настоящее время рассматривается как одна из наиболее тяжелых патологий, ассоциированных с микрососудистым тромбозом. Впервые была описана Eli Moschowitz в 1923 г. Он наблюдал 16-летнюю девушку с лихорадкой, анемией, петехиями, параличом и комой. При аутопсии были обнаружены гиалиновые тромбы в микрососудистом русле. В 1955 г. Gasser и соавт. описали пять детей с острой почечной недостаточностью, сопровождавшейся гемолитической анемией и тромбоцитопенией, и ввели в клиническую практику термин гемолитико-уремический синдром (ГУС).
В настоящее время ТТП и ГУС рассматриваются как проявления тромботической микроангиопатии. Этот патологический процесс был впервые описан Symmers и соавт. в 1952 г. Тромботическая микроангиопатия морфологически проявляется утолщением стенок сосудов микроциркуляторного русла (преимущественно капилляров и артериол), отеком и слущиванием эндотелиальных клеток от базальной мембраны, образованием тромбоцитарных сгустков и частичной или полной обструкцией просвета пораженного сосуда, при этом периваскулярное воспаление не характерно, а тромбы состоят почти исключительно из тромбоцитов. Обструкция просвета сосудов приводит к развитию ишемии и инфарктов органов. Характерным признаком тромботической микроангиопатии является тромбоцитопения и гемолитическая анемия, что связано с потреблением и разрушением тромбоцитов и эритроцитов в микроциркуляторном русле [9,24].
Клинические проявления тромботической микроангиопатии зависят от локализации повреждения микрососудов и, следовательно, от вовлечения в патологический процесс различных органов. Так, ТТП характеризуется пентадой симптомов: тромбоцитопения, микроангиопатическая анемия, лихорадка, поражение почек и неврологическая симптоматика. Неврологические проявления ТТП крайне разнообразны и варьируют от небольших нарушений поведения и затуманенности сознания до выраженных сенсорно-моторных нарушений, афазии, судорог и комы. Для ГУС также характерна тромбоцитопения и микроангиопатическая гемолитическая анемия, но с преимущественным поражением почек. Таким образом, при ТТП преобладают признаки поражения головного мозга, тогда как при ГУС в патологический процесс в основном вовлекаются почки. Кроме того, при ТТП могут наблюдаться боли в животе, панкреатит, гематурия, нарушения сердечного ритма, нарушения зрения. Все эти симптомы обусловлены нарушением микроциркуляции в различных тканях и органах, включая коронарные сосуды, сетчатку, сосуды, кровоснабжающие желудочно-кишечный тракт.
После внедрения терапии свежезамороженной плазмы и значительного снижения летальности у больных ТТП удалось проследить дальнейшую судьбу этих пациентов, и стало очевидным, что этиология ТТП характеризуется значительным разнообразием, как и дальнейшее течение этого заболевания. В настоящее время выделяют наследственную (семейную, врожденную) форму ТТП, которая носит название синдрома Апшою-Шульмана и обусловлена генетическим дефектом протеазы vWF-ADAMTS 13, и приобретенную форму ТТП, обусловленную формированием антител к ADAMTS 13 или ее ингибитора [4]. Мутации в гене ADAMTS 13 вызывают значительное снижение уровней этого фермента в плазме крови или выраженное нарушение его активности. При тяжелом генетически обусловленном дефиците ADAMTS 13 эпизоды ТТП могут начаться с раннего детства, однако у ряда больных заболевание долго может себя не проявлять, вплоть до воздействия какого-либо сильного провоцирующего фактора. Например, триггером для развития первого эпизода ТТП у таких больных может стать беременность, различные инфекционные заболевания и септические состояния, сопровождающиеся массивным выбросом провоспалительных цитокинов, а также прием оральных контрацептивов, так как содержащиеся в них эстрогены стимулируют выброс ультравысокомолекулярных мультимеров vWF из эндотелиоцитов. У ряда больных с выраженным наследственным дефицитом ADAMTS 13 (активность ADAMTS 13 в плазме крови – менее 5-10%) ТТП принимает хроническое рецидивирующее течение с рождения, а ведущим синдромом становится прогрессирующая почечная недостаточность. Впервые двух таких детей описали Шульман в 1960 г. и Апшоу в 1978 г., в честь которых такая рецидивирующая форма ТТП с преимущественным поражением почек у детей стала носить название синдрома Апшоу-Шульмана. При редкой врожденной форме ТТП рецидивы могут возникать каждые 3-4 недели. Такую форму заболевания часто называют хронической рецидивирующей ТТП. У двух третей больных с относительно более распространенной приобретенной формой ТТП в случае успешной терапии повторные эпизоды не возникают, тогда как у трети больных развиваются рецидивы [21]. Когда именно разовьется рецидив, предсказать невозможно. Период ремиссии может длиться от нескольких дней до десятков лет, однако наиболее часто рецидив развивается в течение года после первого эпизода ТТП. Триггером к развитию рецидива могут служить беременность, хирургическое вмешательство, инфекция, вакцинация.
В настоящее время принята следующая классификация тромботических микроангиопатий (см. табл. 1).
Таблица 1. Патологические процессы, ассоциированные с тромбоцитопенией и микроангиопатическим гемолизом.
| Тромботическая микроангиопатия: |
|---|
| Тромботическая тромбоцитопеническая пурпура (ТТП): дефицит активности протеазы фактора фон Виллебранда (vWF) ADAMTS 13 − семейная (врожденная, хроническая рецидивирующая, синдром Апшоу-Шульмана) (дефект гена ADAMTS 13, постоянное снижение активности ADAMTS 13 в плазме крови до 5-10%) − приобретенная (спорадическая) (ингибиторы ADAMTS 13 или аутоантитела IgG к ADAMTS 13, аутоантитела выявляются у 44-94%) (рецидивирующая в трети случаев) Гемолитико-уремический синдром (ГУС) Типичная (эпидемическая) форма: шига-токсин ассоциированный ГУС у детей Атипичные формы: − семейная / наследственная / врожденная /хроническая рецидивирующая форма (дефект фактора Н, компонента комплемента С3) − приобретенная / спорадическая (антитела к компонентам комплемента) Вторичная ГУС/ТТП: − индуцированная лекарственными препаратами (оральные контрацептивы, ингибиторы функции тромбоцитов клопидогрел и тиклопидин, циклоспорин А, митомицин С, такролимус, гемцитабин, комбинированная противоопухолевая терапия) − постинфекционная (S. pneumoniae) − трансплантация костного мозга − лучевая терапия − беременность − системные заболевания (системная красная волчанка, антифосфолипидный синдром, васкулиты) − употребление алкоголя − метастатические опухоли − хирургические вмешательства − острый респираторный дистресс-синдром (ОРДС) − идиопатическая ДВС-синдром |
Молекулярные основы патогенеза тромботической микроангиопатии
Moake с соавт. в 1982 г. впервые выявили аномальные мультимерные комплексы vWF у пациентов с ТТП и сделали предположение о возможной роли vWF в патогенезе ТТП [16]. Характерным признаком ТТП является дефицит плазменной протеазы, расщепляющей мультимеры vWF – ADAMTS 13 (a disintegrin and metalloprotease with thrombospondin type 1 motifs member 13 – дизинтегрин-подобной металлопротеазы с последовательностью 13 по типу тромбоспондина 1). При семейных формах ТТП наблюдается наследственный дефект этого фермента, в то время как приобретенные формы ТТП характеризуются наличием антител-ингибиторов vWF-протеазы [19].
Фактор Виллебранда представляет собой высокомолекулярный мультимер, образующийся при полимеризации мономерных субъединиц с молекулярной массой 225 кДа в эндотелиальных клетках и мегакариоцитах, и накапливающийся в тельцах Weibel-Palade в эндотелиальных клетках и а-гранулах тромбоцитов. Эти ультравысокомолекулярные мультимеры vWF (ULVWF) секретируются активированными эндотелиоцитами наподобие «лент». В норме эти «ленты» высокомолекуляных комплексов vWF сразу же после экспрессии на плазматической мембране подвергаются распаду на фрагменты с Мг 189, 176 и 140 кДа под действием плазматической металлопротеазы ADAMTS 13 и, следовательно, в циркулирующей крови не обнаруживаются [4]. Физиологическая роль мультимера vWF заключается в обеспечении адгезии тромбоцитов к субэндотелиальному матриксу в условиях повреждения сосуда и гемодинамического стресса. Низкомолекулярные фрагменты vWF, циркулирующие в системном кровотоке, обладают слабой способностью к связыванию с тромбоцитами и не проявляют гемостатическую активность. В то время как аффинность отдельных субъединиц vWF к тромбоцитам чрезвычайно мала, мультимеры vWF обеспечивают одновременно множество участков связывания с рецепторами Ib тромбоцитов, что позволяет значительно увеличить силу взаимодействия vWF-тромбоцит. Так, аффинность высокомолекулярной формы vWF к тромбоцитам в 10 раз превышает таковую у отдельных субъединиц vWF [14]. Мультимерные «ленты» ULVWF могут фиксироваться на поверхности мембран эндотелиальных клеток при помощи Р-селектина, который секретируется из телец Weibel-Palade одновременно с ULVWF. В результате в условиях относительного или абсолютного дефицита ADAMTS 13 микрососуды оказываются перекрыты гигантскими ультравысокомолекулярными vWF, на которых оседает все возрастающее количество тромбоцитов, образующих блокирующие микрососудистое русло тромбоцитарные тромбы. Одними из факторов, которые стимулируют выброс ULVWF из эндотелиальных клеток, являются провоспалительные цитокины TNF-альфа и ИЛ-6. В связи с этим состояния, сопровождающиеся активацией процессов системного воспаления, в т.ч. такие осложнения беременности, как преэклампсия, могут стать стимулом к развитию тромботической микроангиопатии.
В норме у здоровых людей активность ADAMTS 13 варьирует в значительных пределах от 50 до 170%. Cнижение активности vWF-протеазы ниже нормы (мене 50%) наблюдается в течение третьего триместра беременности, при циррозе печени, диссеминированных опухолях и воспалительных заболеваниях. У пациентов, переживших ТТП, мультимерные комплексы vWF обнаруживаются лишь в острую фазу заболевания и не обнаруживаются в кровотоке после выздоровления. Возможно, при массивном повреждении эндотелия происходит значительный выброс vWF из гранул; при этом возникает относительная недостаточность металлопротеазы. Однако у пациентов, страдающих рецидивирующей формой ТТП, мультимеры vWF в кровотоке выявляются постоянно: как в острую фазу заболевания, так и в период ремиссии. Такая рецидивирующая форма заболевания чаще является наследственной и обусловлена отсутствием или дефицитом протеазы ADAMTS 13. Так, у большинства пациентов с семейной формой ТТП активность ADAMTS 13 в плазме крови составляет 5-10%, в то время как у большинства пациентов с приобретенной идиопатической ТТП подобное снижение активности ADAMTS 13 выявляется только в период рецидивов [22].
В отличие от сериновых протеаз, у металлопротеазы vWF в норме не обнаруживается плазменный ингибитор. Если для большинства металлопротеаз период полужизни измеряется секундами и минутами, для vWF-протеазы этот показатель составляет 2-4 дня [5]. Поэтому у пациентов с рецидивирующей ТТП и наследственным дефектом vWF-протеазы при применении плазмы, содержащей vWF-протеазу, может быть достигнута ремиссия заболевания. При дефиците vWF, обусловленной наличием ингибитора, целью плазмафереза является удаление патогенных IgG; возможно также применение иммунносупрессивных препаратов (глюкортикоидов, винкристина). Ингибитор vWF-протеазы вновь появляется в крови через 3 месяца после лечения.
Антитела IgG к ADAMTS 13 выявляются у 44-94% пациентов с приобретенной формой ТТП [5]. Их уровень обычно возрастает в острый период или во время рецидива ТТП, тогда как в период ремиссии антитела к ADAMTS 13 в плазме крови у таких больных могут не выявляться. Постоянное обнаружение антител более характерно для больных с частыми рецидивами заболевания, для которых также характерно выявление выраженного дефицита ADAMTS 13. Причинами такого транзиторного выявления антител к ADAMTS 13 может быть, с одной стороны, недостаточная чувствительность доступных в настоящее время диагностических методик, а с другой стороны это может объясняться дефектами иммунной регуляции, на фоне которых возможно возникновение интермиттирующего дефицита ADAMTS 13 под действием различных провоцирующих факторов (беременность, инфекционные заболевания).
Лабораторная диагностика тромботической микроангиопатии
Типичными лабораторными проявлениями ГУТ/ТТП является тромбоцитопения и гемолитическая анемия [6]. Характерно увеличение содержания ЛДГ в сыворотке, что обусловлено активацией гемолиза, а также является признаком тканевой ишемии. Другими признаками гемолиза и перераздражения эритроцитарного ростка служат повышение билирубина (преимущественно непрямого), количества свободного гемоглобина и ретикулоцитов в периферической крови. Характерными признаками микроангиопатической природы гемолиза является обнаружение фрагментов эритроцитов – шистоцитов и отрицательная реакция Кумбса. Причиной образования шистоцитов является резкое сужение сосудов, создающих условия для гемодинамического стресса, обуславливающего фрагментацию эритроцитов. Диагностические подходы, используемые при тромботической микроангиопатии, описаны в таблице 2.
Особенности тромботической микроангиопатии, ассоциированной с беременностью. Новый взгляд на патогенез тяжелых форм гестоза
Беременность по праву считается одним из важнейших триггерных факторов для развития ТТП. В 12-31% случаев ГУС/ТТП развивается во время беременности или в раннем послеродовом периоде [1,12]. Заболеваемость ТТП во время беременности составляет 1 на 25-100 000 [1]. До внедрения в клиническую практику плазмафереза материнская смертность при ГУС/ТТП составляла 95%, а перинатальная – 80% [12]. Во время беременности наблюдается, с одной стороны, прогрессивное повышение уровней vWF, вероятно, под действием эстрогенов, а с другой – снижение активности ADAMTS 13, что может быть обусловлено повышенным потреблением этого фермента, действие которого направлено на разрушение избыточных количеств ультравысокомолекулярных мультимеров vWF, экспрессируемых активированным эндотелиоцитами. Таким образом, беременность может стать провоцирующим фактором для развития ТТП при генетическом дефекте ADAMTS 13. Кроме того, ТТП во время беременности описана и у пациенток с антителами к ADAMTS 13. Был описан интересный клинический случай: у 23-летней женщины в течение 73 месяцев было четыре беременности, заканчивавшиеся самопроизвольными абортами в первом триместре, после чего у нее развивались эпизоды ТТП, регрессировавшие на фоне лечения кортикостероидами и плазмаферезом. После имплантации противозачаточного средства новых эпизодов ТТП не отмечалось [15]. В настоящее время критериями для постановки диагноза из пяти характерных признаков этого заболевания служат только тромбоцитопения и микроангиопатическая гемолитическая анемия. Клинически ТТП/ГУС в этом случае часто бывает трудно отличить от тяжелой формы гестоза, эклампсии и HELLP-синдрома, для которых также характерно развитие тромоцитопении и микроангиопатической гемолитической анемии [23]. Ситуация осложняется еще и тем, что HELLP-синдром и экламптические судороги могут развиваться в отсутствие типичных признаков тяжелого гестоза. Так, по данным Katz V. и соавт. (2000), у 60% женщин эклампсия развилась на фоне нормального артериального давления и была первым проявлением гестоза [8].
Таким образом, развитие тромботической микроангиопатии характерно и для HELLP-синдрома, тромботической тромбоцитопенической пурпуры, гемолитико-уремического синдрома, а также является одним из проявлений катастрофического антифосфолипидного синдрома (КАФС). Это свидетельствует о едином механизме патогенеза этих заболеваний. Известно, что АФС ассоциируется с высокой частотой развития таких патологий беременности, как СЗРП, внутриутробная гибель плода, преждевременные роды, гестозы. Кроме того, рядом исследователей описаны случаи возникновения HELLP-синдрома у женщин с АФС, что лишний раз подтверждает роль патологии гемостаза как предрасполагающего фактора к возникновению HELLP-синдрома. Koenig и соавт. (2005) описали женщину с АФС, у которой беременность осложнилась развитием HELLP-синдрома, а после оперативного родоразрешения развилась клиническая картина КАФС с инфарктами печени, ЖКТ и костного мозга вследствие прогрессирующей микроангиопатии [12]. Следует также учитывать, что HELLP-синдром может быть первым проявлением АФС. Появились данные о роли антител к ADAMTS 13 в качестве причины тромбоцитопении у больных СКВ, что может быть одним из критериев неблагоприятного прогноза заболевания у таких пациентов [10] Pourrat O. и соавт. (2013) описывают взаимосвязь между дефицитом ADAMTS 13 и развитием HELLP-синдрома [18]. Более того, появились интересные данные о том, что антитела к ADAMTS 13 могут формироваться в условиях антифосфолипидного синдрома, что может являться важнейшим фактором развития тромботических и акушерских осложнений [1]. Таким образом, антитела к ADAMTS 13 и дисфункция ADAMTS 13 может развиваться и при других аутоиммунных заболеваниях, помимо приобретенной ТПП, в частности, в условиях АФС.
Согласно нашей концепции, наследственный и/или приобретенный дефицит ADAMTS 13, наряду с генетический тромбофилией и антифосфолипидным синдром, включая катастрофическую его форму, должен рассматриваться как один из значимых патогенетических факторов возникновения критических состояний в акушерстве (см. рис. 1). Все женщины, перенесшие тяжелые формы преэклампсии, HELLP-синдром, преждевременную отслойку нормально расположенной плаценты, септический шок, геморрагический шок, должны быть обследованы на выявление активности ADAMTS 13 и наличия ее ингибиторов. В случае отягощенного анамнеза и выявления нарушений в системе ADAMTS 13 оценка уровней этого фермента и титров его ингибитора в течение беременности в динамике позволит оценить риски развития повторных тяжелых тромботических и акушерских осложнений и своевременно принять вопрос о дальнейшей тактике ведения пациентки и необходимости в досрочном родоразрешении.
 | Рисунок 1: Осложнения беременности как отдельная форма тромботической микроангиопатиии. |
Более того, в последнее время появляются новые данные о роли дефицита ADAMTS 13 в патогенезе различных инфекционных заболеваний и тромботических осложнений, включая острый инфаркт миокарда, ишемический инсульт, гипертензивные кризы, сепсис, малярия (см. рис. 2).
 | Рисунок 2: Состояния, ассоциированные с дефицитом ADAMTS 13. |
Принципы терапии ГУС/ТТП
Терапией выбора при ТТП/ГУС является применение свежезамороженной плазмы или плазмаферез. Применение обменного переливания плазмы позволяет снизить уровень смертности при ГУС/ТТП с 80 до 10%. Впервые эффективность обменного переливания плазмы была показана у пациента с ТТП Rubinshtein в 1959 г. Целью применения плазмафереза является возмещение уровня vWF-протеазы, удаление антител, блокирующих активность ADAMTS 13, провоспалительных цитокинов, компонентов комплемента из системного кровотока, а также возмещение дефицита естественных антикоагулянтов, что особенно важно при сочетании ТТП/ГУС с генетическими тромбофилиями и АФС. При наследственной форме, обусловленной гомозиготной или двумя гетерозиготными мутациями ADAMTS 13, эффективно применение свежезамороженной плазмы, а для профилактики рецидивов заболевания переливания плазмы необходимо применять раз в 2-3 недели [2]. У больных с приобретенной формой ТТП, у которых дефицит ADAMTS 13 в большинстве обусловлен не абсолютным отсутствием этого белка, а блокадой его активности вследствие циркуляции аутоантител, только переливания свежезамороженной плазмы может оказаться недостаточным, так как имеющиеся антитела будут блокировать и вновь поступающие в организм количества ADAMTS 13. Тем не менее, инфузия свежезамороженной плазмы у больных с приобретенной формой ТТП должна быть начата сразу же после появления подозрения ТТП при отсутствии возможности начать плазмаферез в экстренном порядке или до того, как будет уточнен диагноз [20].
На сегодня для лечения ГУС/ТТП, помимо терапии плазмой, применяется целый ряд методов и лекарственных препаратов, однако эффективность большинства их них остается недоказанной (см. табл. 3). Эти методы направлены на подавление синтеза аутоантител и применяются у больных с приобретенной ТТП и аутоантителами к ADAMTS 13 при отсутствии эффекта от стандартной терапии с применением плазмафераза и свежезамороженной плазмы. Возможные варианты терапии включают высокие дозы глюкокортикоидов, ритуксимаб (моноклональное антитела к CD20 на В-лимоцитах), в комбинации с циклофосфамидом, циклоспорин, спленэктомию. Так, появились данные о повышении эффективности терапии приобретенной ТТП при одновременном применении плазмафереза и глюкокортикоидов [2,11]. Рекомендуют начинать преднизолон внутривенно в дозе 200 мг в день сразу же после установления диагноза ТТП и продолжать лечение с постепенным снижением дозы вплоть до выздоровления [13].
Таблица 3. Методы лечения тромботической микроангиопатии.
| Лечение | Способы применения и дозировка | Показания |
|---|---|---|
Лечение с доказанной эффективностью | ||
Плазмаферез | Переливание 1-2 (60-80 мл/кг/день) доз плазмы в день | Терапия выбора при ГУС/ТТП взрослых, жизненно необходима при поражении ЦНС, восстанавливает функцию почек; риск перегрузки объемом при переливании плазмы отсутствует даже у пациентов с сердечной и почечной недостаточностью Начальная терапия при всех вариантах тромботической микроангиопатии Терапия выбора при аутоиммунной ТТП |
Свежезамороженная плазма | 30-40 мл/кг в первый день, далее – по 10-20 мл/кг/день | Терапия выбора, если плазмаферез недоступен; эффективна для профилактики и лечения рецидивов ГУС/ТТП При семейной форме применяется для профилактики рецидивов каждые 2-4 недели Механизм действия – замещение недостающего фермента ADAMTS 13 |
Криопреципитированная плазма (очищенная от мультимеров vWF, фибриногена, фибронектина) | 30-40 мл/кг в первый день, далее – по 10-20 мл/кг/день | Терапия второй линии при неэффективности свежезамороженной плазмы и плазмафереза |
Спленэктомия | Частые рецидивы ГУС/ТТП, ТМА, рефрактерная к терапии. Механизм действия неизвестен, удаление клеток памяти? | |
Глюкокортикоиды | 1-2 мг/кг/день | Дополнительная терапия при недостаточной эффективности применения свежезамороженной плазмы/плазмафереза при ТМА, связанной с антителами |
Гамма-глобулин | 400 мг/кг/день в/в | Эффективность не доказана |
Иммуномодуляторы (винкристин, циклофосфамид, циклоспорин) | Механизм действия – иммуносупрессия Дополнительная терапия при недостаточной эффективности применения свежезамороженной плазмы/плазмафереза при ТМА, связанной с антителами | |
Антитромбоцитарные препараты | Ацетилсалициловая кислота 100-300 мг/сут. Клопидогрел 75-150 мг/сут. | Эффективность не доказана, увеличивают риск кровотечений ТМА с выраженными ишемическими органными поражениями |
Исследуемые препараты | ||
Рекомбинантный ADAMTS 13 | При врожденной форме ТМА замещение дефицитного фактора При аутоиммунной форме преодоление эффекта ингибитора ADAMTS 13? | |
Каплацизубам, ARC1779, ARC 15105 | Блокирование доменов vWF A1, конкурирование за связывания с тромбоцитарным GP Ib/IX | |
Важно отметить, недавно появившиеся новые данные о том, что тяжелые формы гестоза являются проявлением тромботической микроангиопатии, могут свидетельствовать о том, что плазмаферез и переливание свежезамороженной плазмы должны рассматриваться в качестве терапии выбора у таких больных. Данные исследований Isler и соавт. (2001) свидетельствуют о том, что применение глюкокортикоидов до и после родов способствует уменьшению тяжести HELLP-синдрома, потребности в гемотрансфузии и позволяет продлить беременность на 24-48 ч, что важно для профилактики респираторного дистресс-синдрома новорожденных [7]. Предполагается, что применение глюкокортикоидов может способствовать восстановлению функций эндотелия, блокаде аутоиммунных патогенетических механизмов, предотвращать внутрисосудистое разрушение эритроцитов и тромбоцитов и прогрессирование ССВО. В частности, рассматривая HELLP-синдром как вариант приобретенной тромботической микроангиопатии, эффективность глюкокортикоидов можно объяснить блокирующим эффектом в отношении антител к ADAMTS 13 и антифосфолипидных антител. Однако вслед за улучшением клинической картины, отмечаемым в течение 24-48 ч на фоне применения глюкокортикоидов, может возникнуть так называемый ребаунд-эффект, проявляющийся ухудшением состояния беременной. Таким образом, введение глюкокортикоидов не предотвращает полностью развитие патологического процесса, а лишь кратковременно улучшает клиническую картину, создавая условия для более успешного родоразрешения.
Важно отметить, что всем пациентам с ТТП, несмотря на выраженную тромбоцитопению, необходимо проведение тромбопрофилактики низкомолекулярным гепарином. Даже при тяжелой тромбоцитопении переливание тромбоцитов показано только у пациентов с жизнеугрожающими кровотечениями, так как это может спровоцировать прогрессирование тромботической микроангиопатии.
Открытия последних лет, связанные с изучением молекулярных механизмов тромботический микроангиопатии, позволяют сделать вывод о том, что эта патология является гораздо более распространенной, чем было ранее принято считать. Важнейшим триггером к развитию как тромботической тромбоцитопенической пурпуры, так и других вариантов тромботической микроангиопатии является беременность. Причинами тому может являться физиологическое повышение уровней фактора фон Виллебранда, характерное для беременности, активное потребление и истощение запасов ADAMTS 13, что может усугублять ранее скрытый, умеренный генетически обусловленный дефицит этого фермента. Кроме того, последние данные указывают на взаимосвязь между циркуляцией антифосфолипидных антител и приобретенным дефицитом ADAMTS 13. В частности, такие механизмы могут играть важную роль в патогенезе тяжелых плацентарных осложнений беременности, в т.ч. преэклампсии, HELLP-синдрома, преждевременной отслойки нормально расположенной плаценты, быть триггером в развитии септического шока. В настоящее время критерии для установления диагноза тромботической микроангиопатии значительно расширены. Эту патологию следует исключать у всех пациентов, у которых выявляется тромбоцитопения в сочетании с гемолитической анемией. Своевременная диагностика тромботической микроангиопатии имеет колоссальное значение для выбора тактики лечения, в т.ч. может полностью изменить подход к терапии пациенток с тяжелыми формами преэклампсии и HELLP-синдромом. Обследование пациенток на дефицит ADAMTS 13 и определение уровней ингибитора этого фермента позволяет пролить свет на патогенез ранее перенесенных пациентками тромботических и тяжелых акушерских осложнений и планировать специфическую профилактику во время последующих беременностей. Согласно нашей концепции у всех женщин, перенесших тяжелые формы преэклампсии, HELLP-синдром, преждевременную отслойку нормально расположенной плаценты, септический шок, геморрагический шок, артериальные и венозные тромботические осложнения, необходимо проводить исследование на активность и на выявление ингибиторов ADAMTS 13. В случае отягощенного акушерского и тромботического анамнеза и выявления нарушений в системе ADAMTS 13 определение уровней этого фермента и титров его ингибитора в течение беременности в динамике позволит оценить риски развития повторных осложнений и своевременно принять вопрос о дальнейшей тактике ведения пациентки и необходимости в досрочном родоразрешении.


