Что такое трисомия?
Статья опубликована: 2018-12-30
Рейтинг: 5 из 5
Если взять нормального человека и рассмотреть его геном, то мы обнаружим 23 пары хромосом. По одной хромосоме из пары мы получаем от мамы и папы. Но порой могут возникать нарушение в их числе. Так образуются утроения — трисомии. Это очень тяжелые генетические заболевания, которые зачастую не совместимы с жизнью. Если несколько хромосом имеют аномалии, то плод скорее всего не доживет до рождения или умрет на первых днях жизни.

Основные виды трисомий
Если мутация произошла только в одной хромосоме, то такие дети выживают, но становятся инвалидами на всю жизнь. О них приходится все время заботится и в конце концов годам к 40 они погибают. Рассмотрим основные виды трисомий:

Проблемы с сердцем и мочеполовой системой
Диагностировать трисомии возможно уже в первом триместре, а именно начиная с 9-ой недели беременности. Женщина сдает кровь из вены, далее из её крови выделяется ДНК плода для исследования. Такой анализ называется неинвазивный пренатальный тест. Генетический центр «ДТЛ» проводить НИПТ-тесты уже более 4 лет. Мы рекомендуем сделать неинвазивное исследование всем беременным женщинам, особенно после 35 лет.
Пренатальный скрининг; хромосомные аномалии
Частота врожденных пороков развития составляет 2-3%, еще 5% новорожденных имеют так называемые малые аномалии. Причинные факторы их являются гетерогенными и включают хромосомные аномалии, моногенные заболевания, влияние тератогенов, материнские заболевания (инсулинзависимый сахарный диабет, фенилкетонурия), инфекции (краснуха, цитомегалия и др). Но большинство врожденных аномалий развития является мультифакториальными, т.е. зависят от комбинации генетических факторов и воздействия агрессивных факторов внешней среды.
Что такое пренатальный скрининг
Пренатальный скрининг, диагностика и лечение является относительно новой проблемой в акушерстве. Началом пренатального скрининга была, возможно, эра ультразвуковой диагностики в акушерстве, которая началась около двух десятилетий назад. С открытием новых генов и их фенотипов становится все более возможным пренатальный генетический диагноз. Следует различать понятия скрининга и диагностики.
Пренатальный скрининг позволяет выявить индивидов высокого риска осложнений среди популяции индивидов с низким риском осложнений. Специфичность и чувствительность скрининговых тестов очень важны, учитывая возможность ложноположительных и ложноотрицательных результатов скрининга.
Пренатальная диагностика, конечно, более специфическая, чем скрининг (например, амниоцентез или биопсия хориона), но имеет и больший риск осложнений. Первым шагом по определению риска для плода является скрининг матери о наличии определенных состояний или заболеваний.
Нередко возникает вопрос о вероятности роста частоты врожденных пороков у потомков семейных пар, которые получали лечение по поводу бесплодия. Тяжелая олигоспермия и азооспермия ассоциируются со сбалансированными транслокациями хромосом (3-5%), синдромом Кляйнфельтера (47, ХХУ), аномалиями и микроделеции У-хромосомы.
Аномалии Х-хромосомы (ХХУ, ХХХ, Х-мозаицизм при синдроме Тернера) ассоциируются с пониженной фертильностью (субфертильностью), а также увеличением риска хромосомных аномалий у потомков. В 2/3 пациентов с врожденным отсутствием семявыносящих протоков имеет место хотя бы одна мутация гена, который отвечает за развитие кистозного фиброза. Итак, эти пациенты подлежат скринингу на наличие кистозного фиброза. Таким пациентам обычно показана интрацитоплазматическая инъекция сперматозоида в яйцеклетку, хотя наличие мутантного гена по кистозному фиброзу может влиять на репродуктивные намерения.
Хромосомные аномалии
Старший возраст матери является фактором риска хромосомных аномалий в связи с увеличением возможности нерасхождения хромосом в процессе мейоза. Фертилизация гаметы с одной лишней хромосомой приводит к образованию продукта оплодотворения с 47 хромосомами. Следовательно, растет частота анеуплоидии — количества хромосом в продукте оплодотворения, большей или меньшей 46. Нерасхождение хромосом может иметь место в аутосомах (трисомия 21, 13, 18) или половых хромосомах (моносомия 45, Х, или трисомия 47, ХVV, 47, ХХХ и др). Несбалансированные транслокации хромосом сопровождаются аномальным количеством хромосомного материала (целой хромосомы или ее части). Риск для ребенка зависит от типа транслокации.
Факторы риска рождения детей с хромосомными аномалиями
Частота хромосомных аномалий у живых новорожденных составляет 0,5%, у мертворожденных — 5%, у абортусов при самопроизвольных выкидышах — 50%. Частой хромосомной аномалией является анеуплоидия — увеличение или недостаток одной хромосомы. У рожденных живыми наиболее часто встречаются такие хромосомные аномалии, как трисомия 21 (1: 800), трисомия 18 и трисомия 13.
Трисомия 16 наиболее часто приводит к самопроизвольным выкидышам, а в случае трисомии 18 в большинстве случаев имеет место мертворождение. При наличии в анамнезе трисомии у плода риск рецидива при повторной беременности составляет 1%. В случае триплоидии обычно имеет место самопроизвольный аборт или гестационная трофобластическая болезнь. В редких случаях ребенок может родиться с триплоидией, но продолжительность жизни не превышает 1 года.
Хромосомные аномалии часто сопровождаются выраженными фенотипическими проявлениями и врожденными пороками развития, хотя их не всегда можно обнаружить при ультразвуковом скрининге.
Наиболее точным методом диагностики хромосомных аномалий является исследование кариотипа плода. Для некоторых хромосомных синдромов (синдром Дауна) существуют скрининговые тесты, например тройной тест:
1) уровень а-фетопротеина;
3) уровень в-субъединицы ХГЧ в сыворотке крови матери.
Синдром Дауна
Трисомия 21 или избыточная 21 хромосома, вызывает синдром Дауна. Наличие такого кариотипа часто является причиной самопроизвольного выкидыша или мертворождения. Но, несмотря на «пренатальные фильтры», ежегодно рождаются несколько тысяч детей с синдромом Дауна. Риск этого заболевания имеет значительную зависимость от увеличения возраста родителей.
Типичный фенотип синдрома Дауна включает низкий рост, плоское лицо, эпикант, гипертелоризм, поперечную складку ладоней, задержку физического и умственного развития, хотя коэффициент интеллекта (10) может колебаться от 40 до 90. Ассоциированные аномалии представлены пороками развития сердца, дуоденальной атрезией или стенозом, короткими конечностями. В 50% случаев синдром Дауна можно заподозрить при ультразвуковом обследовании, но низкая чувствительность не позволяет использовать УЗИ как скрининговый метод. Современный скрининг синдрома Дауна заключается в использовании тройного теста: АФП (снижение), эстриол (снижение) и ХГЧ (повышение) в сроке 15-19 нед гестации.
Трисомия 18
Трисомия 18 (синдром Эдвардса) — вариант трисомии, который также может быть обнаружен при использовании тройного теста. Этот синдром является несовместимым с жизнью пациентов старше 2 лет и нередко является причиной антенатальной или неонатальной смерти. Синдром Эдвардса сопровождается многочисленными врожденными аномалиями, которые могут быть обнаружены при УЗИ, делает возможным ультразвуковой скрининг.
Типичными признаками синдрома Эдвардса есть сжатые кулаки, искривленные пальцы и др. Ассоциированные с этим синдромом врожденные аномалии включают пороки сердца (дефект межжелудочковой перегородки, тетрада Фалло), омфалоцеле, диафрагмальную грыжу, дефекты нервной трубки, кисты хориоидного сплетения в боковых желудочках мозга и др.
Трисомия 13
Трисомия 13 (синдром Патау) имеет много черт, подобных трисомии 18. Около 85% этих детей умирают до 1 года жизни. Ассоциированные аномалии включают голопрозэнцефалию, щель губы и неба, кистозный гигром, аномальную форму или отсутствие носа, омфалоцеле, гипоплазию левых отделов сердца, аномалии конечностей (сжатые кулаки и согнуты ступни), полидактилию, чрезмерное сгибание пальцев.
В случае синдрома Патау тройной тест не всегда является чувствительным, но эта аномалия обычно может быть диагностирована при рутинном ультразвуковом скрининге.
Робертсоновские транслокации — это соединение между двумя акроцентрическими хромосомами (13, 14, 15, 21, 22) в области их центромеры, что создает новую хромосому. Носители таких хромосом обычно генетически сбалансированными, но существует риск несбалансированных транслокаций у потомков, а также риск самопроизвольных выкидышей. Робертсоновские транслокации хромосомы 21 приводит к развитию синдрома Дауна (риск составляет 10-15%, если носителем транслокации является мать, и 1-2% — если отец).
Реципрокные транслокации возникают при разрыве хромосом и обмене сегментами между двумя различными хромосомами. Сбалансированные носители имеют нормальное количество хромосом (46), тогда как в их потомков увеличивается риск несбалансированных транслокаций, делеций и дупликаций хромосомного материала, что приводит к аномальному фенотипу.
Микроделеции хромосом — субмикроскопические делеции хромосом, которые в некоторых случаях могут быть обнаружены только при использовании молекулярной диагностики и РИ8Н-анализа, но иногда приводят к развитию выраженных клинических синдромов. Скрининг таких состояний рекомендуется при наличии в анамнезе рождения детей с микроделецией хромосом.
Цитологическая диагностика
Флюоресцентная гибридизация используется с целью выявления субмикроскопических делеций и дупликаций хромосом, которые являются слишком маленькими для идентификации методами конвенциональной цитогенетики. Ее также используют для идентификации «мягких» транслокаций и маркерных хромосом. РИ8Н-анализ выполняется на препарате метафазных хромосом, выделенных из культивируемых лимфоцитов, амниоцитов, ворсинок хориона, интерфазных ядер из крови, тканей, ворсинок хориона, амниотической жидкости при пренатальном выявлении аномалий развития плода и необходимости скрининга на анеуплоидии (трисомия 21 и т.д.). РИ8Н-анализ применяется с целью преимплантационной генетической диагностики по выявлению сбалансированных транслокаций или делеций. РИ8Н-метод предоставляет информацию по анализу специфической хромосомы или нескольких хромосом, но не выполняется для определения кариотипа.
Аномалии половых хромосом
Наиболее частыми аномалиями половых хромосом является синдром Тернера (моносомия Х, или 45, Х0, мозаицизм) и синдром Кляйнфельтера (47, ХХУ). Это может быть обусловлено тем обстоятельством, что кариотипы 47, ХХХ и 47, ХVV не имеют выраженных фенотипических различий и поэтому идентифицируются реже.
Индивиды с синдромом Тернера имеют женский фенотип, дисгенезию гонад, низкий рост, первичные, отсутствие вторичных половых признаков, крыловидные складки шеи, низко расположенные уши, низкую заднюю границу роста волос, дискообразную грудную клетку с широкой расстоянием между сосками, аномалии почек, лимфедему конечностей при рождении и кардиоваскулярные аномалии, чаще коарктацию аорты. Но при ультразвуковом исследовании может проявляться только одна аномалия — кистозная гигрома. Скрининг-теста синдромом Тернера еще не существует, и поэтому частота рецидивов не определена.
В случае синдрома Кляйнфельтера развитие яичек сначала является нормальным. Но присутствие не менее 1 лишней Х-хромосомы приводит к гибели зародышевых клеток на этапе их поступления в мейоз, что приводит к уменьшению яичек и гиалинизации семенных протоков. Итак, классические симптомы синдрома Кляйнфельтера включают бесплодие, гинекомастию, задержку умственного развития, повышения уровня гонадотропинов вследствие уменьшения уровня циркулирующих андрогенов. Скрининг-теста по выявлению синдрома Кляйнфельтера также не существует, следовательно, пренатальный диагноз этих хромосомных аномалий возможно только при использовании биопсии хориона или амниоцентеза.
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
Показания для тестирования на ломкую Х-хромосому
Геномный импринтинг — процесс, при котором активация гена происходит преимущественно в материнской или преимущественно в родительской хромосоме, но не в обеих хромосомах. Нормальное развитие имеет место лишь в том случае, если присутствуют обе копии (материнская и отцовская) импринтинг-ген. Импринтинг-ген неактивен, значит, активный ген теряет (путем делеции) или получает мутацию, в таком случае плод будет пораженным. Лишь несколько генов могут испытывать импринтинга.
Примерами геномного импринтинга может быть синдром Ангельмана и полный пузырный занос (вариант гестационной трофобластической болезни).
Синдром Ангельмана характеризуется тяжелой задержкой умственного развития, атаксической походкой, типичным лицом, пароксизмами смеха и судорогами. Ген синдрома Ангельмана является активным только в материнской унаследованной хромосоме, следовательно, если происходит делеция материнской хромосомы 15 или материнская копия гена имеет мутацию, белковый продукт не образуется и плод будет пораженным.
Синдром Ангельмана также может развиться, если обе копии хромосомы 15 является унаследованными от отца (отсутствие материнской копии хромосомы 15). Это состояние получило название унипарентальной дисомии. Унипарентальная дисомия возникает чаще вследствие потери хромосомы у эмбриона с трисомией или добавления хромосомы у плода с моносомией по этой хромосомой. Каждая из хромосом может быть генетически различной (гетеродисомия) или идентичной (изодисомия), в зависимости от времени возникновения этого феномена — в течение первого или второго мейотического деления, соответственно.
Полный пузырный занос обычно является диплоидным (46, ХХ или Х ¥), но может иметь полностью отцовское происхождение, без материнского хромосомного материала. При таких условиях плод не может развиваться. Полный пузырный занос может сопровождать нормальную многоплодную беременность, но в этом случае возрастает риск материнских осложнений (гипертиреоидизм, преэклампсия, преждевременные роды). В отличие от полного пузырного заноса, частичный пузырный занос обычно является триплоидным (69, ХХХ, 69, ХVV), с дополнительным набором отцовских хромосом.
Триплоидия с дополнительным набором материнских хромосом имеет место при ЗВУР плода, врожденных пороках развития и маленькой плаценте.
Митохондриальное наследование
Митохондрии в цитоплазме яйцеклетки (но не сперматозоида) передаются от матери к ее потомкам. Митохондрия имеет собственную ДНК. Существует несколько генетических заболеваний, вызванных мутациями митохондриальной ДНК, — наследственная оптическая нейропатия Лебера, болезнь Ли (подострая некротизирующая энцефаломиелопатия), миоклоническая эпилепсия с «зазубренными красными волокнами». Экспрессия этих заболеваний является вариабельной.
Трисомия 16 хромосомы что это
Выявлена причина 41% выкидышей, которая в противном случае осталась бы невыясненной
Результаты дают прогноз для следующей беременности, позволяют избежать других неинформативных методов обследования или неоправданного применения лекарственных препаратов
Недостатки существующих методов
Ограничением традиционного кариотипирования является клеточная культура, которую нужно выращивать в лабораторных условиях в течение 2-4 недель.
Дегенерирующие, умирающие или уже мертвые клетки для проведения традиционного кариотипирования не подходят, так как их рост в лабораторных условиях зачастую невозможен. Именно это влечет за собой 10-40% риска неудачного результата.
Особенно это касается эмбрионального материала, когда клетки плода погибают при заборе, транспортировке и хранении.
Для Молекулярно-генетических исследований при невынашивании не нужны живые клетки. Достаточно небольшого количества ДНК, и ее всегда можно выделить из полученного материала.
Генетическое исследование ворсин хориона это:
Что предлагает лаборатория «Геномед»?
«Оптима»
Прерывание беременности по медицинским показаниям / внутриутробная гибель плода на поздних сроках беременности
Выполняется 10 дней
«Оптима расширенный»
Неразвивающаяся беременность, выкидыш, прерывание беременности по медицинским показаниям
То же, что Оптима, плюс 13 видов моногенной патологии:
*Определение точечных мутаций невозможно при наличии триплоидии и сильной контаминации образца
Выполняется 10 дней
«Фертус»
Прерывание беременности по медицинским показаниям / внутриутробная гибель плода на поздних сроках беременности
Выполняется 90 дней
Классическое кариотипирование, в среднем, в 20% cлучаев не дает результата, так как культивирование дегенерирующих клеток абортивного материала, в лабораторных условиях затруднительно.
Риск получения ложного результата исследования при применении таких методов, обусловлен контаминацией материнскими клетками при процедуре забора абортивного материала и полиплоидизацией при выращивании клеточной культуры.
У этих проблем теперь есть решение – «Молекулярное кариотипирование абортивного материала ФЕРТУС»
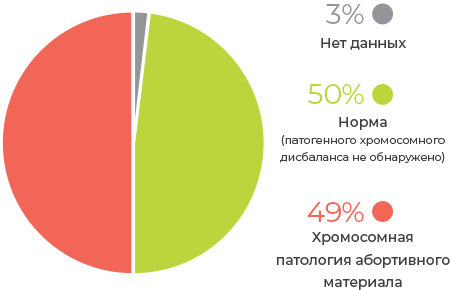
«Молекулярное кариотипирование абортивного материала ФЕРТУС» по методу NGS зарекомендовал себя как высокоэффективный современный тест для диагностики анеуплоидий и может быть предложен для исследования клеток неразвивающегося хориона.

Структура хромосомной патологии, выявленной методом NGS в клетках неразвивающегося хориона
«Молекулярное кариотипирование абортивного материала Фертус» исследование первой линии при потере беременности малого срока, так как метод NGS:
Позволяет детектировать как численные изменения хромосом, так и субмикроскопические вариации числа копий генов (CNV)
Позволяет заподозрить сбалансированные транслокации у родителей
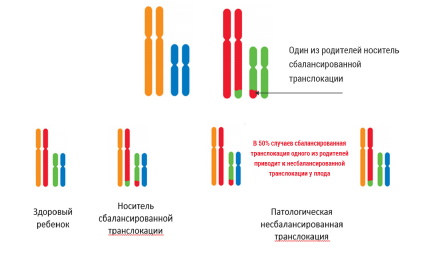
В некоторых случаях выявление носительства сбалансированных хромосомных аномалий у родителей возможно только после проведения теста при использовании плодного материала
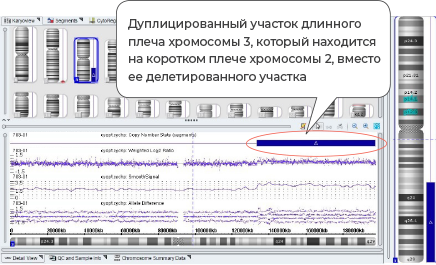
Несбалансированная транслокация между короткими плечами 2 и 3 хромосомы. Риск повторного возникновения данной аномалии 50%
Молекулярное кариотипирование «Оптима» позволяет определить
полную или частичную молярную беременность
Пузырный занос (наиболее частый тип гестационной трофобластической болезни) с полной отцовской однородительской дисомией в 20% и частичный диандрический пузырный занос в 5% случаев может принимать злокачественное течение, поэтому важно вовремя установить его происхождение.
 Полногеномная ОРД отцовского происхождения (полный пузырный занос) – полное отсутствие гетерозиготных вариантов во всех хромосомах
Полногеномная ОРД отцовского происхождения (полный пузырный занос) – полное отсутствие гетерозиготных вариантов во всех хромосомах
Молекулярное кариотипирование «Оптима» также позволяет установить происхождение триплоидии и оценить риски возможных осложнений
Несмотря на результативность молекулярного кариотипирования, 50% причин невынашивания беременности не детектируются, так как не связаны со структурными и количественными изменениями хромосом и в некоторых случаях имеют моногенную природу.
Основные моногенные заболевания как причина спонтанных абортов:
Для детекции всех возможных генетических причин спонтанных абортов лаборатория «Геномед» предлагает исследование «Полное секвенирование генома плодного материала «Фертус»
Технология NGS (Next Generation Sequencing), лежащая в основе WGS (Whole Genome Sequencing), обладает диагностическим преимуществом, так как позволяет детектировать:
Показания к проведению исследования:
Все делеции и дупликации анализируются врачом-генетиком с использованием реферируемых и постоянно пополняемых баз данных.
Для исследования необходимо 20-30 г материала эмбрионального происхождения, который необходимо поместить в специальный набор или в контейнер с 0,9%-ным физиологическим раствором.
Для сбора материала рекомендуется использовать специальные наборы, которые содержат контейнер с консервирующим раствором, специальную упаковку и инструкцию.
Если прерывание беременности произошло:
Многие пациентки, у которых был выкидыш, хотят знать его причину. Тест
«Оптима» обеспечивает точные и полные результаты, которых заслуживаете
Вы и Ваши пациенты.
Если обнаружена
анеуплоидия Если обнаружены
микроделеции/микродупликации Если обнаружена
триплоидия Если обнаружена
однородительская дисомия Определение контаминации
материнскими клетками
Если обнаружена анеуплоидия
Анеуплоидии (моносомии или трисомии одной или нескольких хромосом) встречаются наиболее часто.
Как правило, это случайные события и риск их повторения при следующих беременностях является общепопуляционным. Прогноз благоприятный.
Распределение выявленных анеуплодий
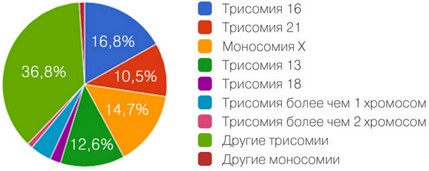
Структура анеуплодий, выявленных в лаборатории «Геномед»
(всего исследовано 303 образца, анеуплодии обнаружены в 95 случаях).
Если обнаружены микроделеции/микродупликации
Микроделеции и микродупликации представляют собой, соответственно, потерю или удвоение хромосомного материала, которые невозможно определить с помощью обычного анализа кариотипа. Несмотря на их малые размеры, подобные перестройки могут приводить к возникновению тяжелых врожденных пороков развития.
При обнаружении микроделеций/микродупликаций в продуктах оплодотворения необходимо направить пациентку на консультацию к врачу-генетику, который оценит клиническое значение перестроек.
Хотя вероятность повторного возникновения аналогичной микроперестройки у плода при следующей беременности крайне мала, подобные случаи описаны, что служит основанием для рекомендаций по проведению инвазивной пренатальной диагностики и молекулярного кариотипирования.
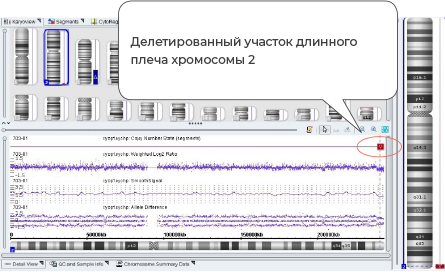
Пациент М., женщина 34 года. МВПР и внутриутробная задержка развития плода. Прерывание беременности на 22 неделе. В результате хромосомного микроматричного анализа была обнаружена микроделеция участка длинного плеча 2 хромосомы и микродупликация участка короткого плеча 17 хромосомы. Молекулярный кариотип: ап2д37.3(240,385,38 1-242, 783,384)х1,17р13.Зр13.1(525-8,350,996)х3
Сочетание микроделеции и микродупликации является признаком несбалансированной транслокации, причиной которой может быть сбалансированная транслокация у одного из родителей.
Результатом сбалансированной транслокации у одного из родителей могут быть повторные неразвивающиеся беременности или рождение тяжелобольного ребенка с хромосомной патологией.
Если обнаружена триплоидия
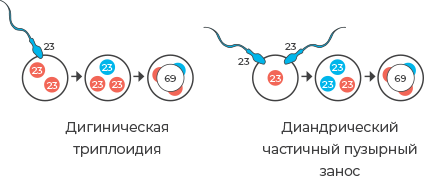
Молекулярное кариотипирование «Оптима» позволяет дифференцировать доброкачественную дигиническую триплоидию, когда дополнительный набор хромосом имеет материнское происхождение от диандрического частичного пузырного заноса, при котором дополнительный набор хромосом имеет отцовское происхождение.
При выявлении триплоидии необходимо рекомендовать молекулярное кариотипирование родителей для установления происхождения дополнительного набора хромосом
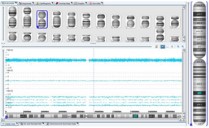
Пациент С., женщина, 31 год. Хромосомный микроматричный анализ. Определяется дополнительный сигнал от полиморфных маркеров на каждой хромосоме. Молекулярное кариотипирование родителей подтвердило отцовское происхождение триплоидии.
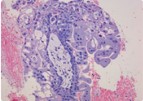
Третичные ворсины хориона циркулярно окружены промежуточным трофобластом с признаками пролиферации. Окраска гематоксилином и зозином, х200.
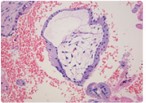
Умеренная гидратация стромы ворсины. Окраска гематоксилином и зозином, х200.
Если обнаружена однородительская дисомия
Полные однородительские дисомии имеют отцовское происхождение и являются характерным признаком пузырного заноса. Проведение молекулярного кариотипирования всегда может определить это и помочь в диагностике, особенно при неоднозначных гистологических данных.
ОБНАРУЖЕНИЕ
ХРОМОСОМНЫХ ПРИЧИН ПУЗЫРНОГО ЗАНОСА
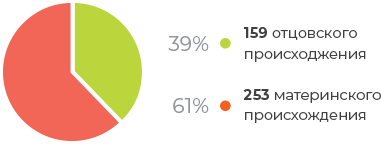
Пузырный занос несет в себе огромный риск для матери. Пузырный занос с полной отцовской однородительской дисомией (ОРД) несет в себе 20%-ный риск возникновения гестационной трофобластической болезни (ГТБ), а при частичном пузырном заносе с триплоидией по отцовской линии шанс возникновения ГТБ равен 5%.
По этой причине всем женщинам с пузырным заносом следует контролировать уровень ХГЧ в крови и наблюдаться у врача после беременности.
Выявленная ГТБ лечится с помощью химиотерапии.
Установление родительского происхождения триплоидии позволяет выдать рекомендации по контролю ГТБ.
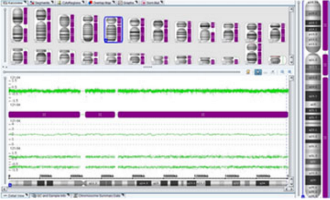
Полногеномная однородительская дисомия отцовского происхождения (полный пузырный занос)

При отсутствии возможности определения отцовского происхождения триплоидии в 61 % случаев будут предприниматься ненужные меры предосторожности для контроля риска ГТБ.
Триплоидия материнского происхождения не связана с пузырным заносом.
Определение контаминации материнскими клетками
Плодный материал всегда содержит примесь материнской крови или тканей (контаминация). Часто такие материнские клетки растут в культуре лучше, чем клетки плода, что может являться причиной ложных результатов.
В результате исследования продуктов оплодотворения в 51% случаев кариотип 46,ХХ был получен при анализе хромосом клеток материнского происхождения.
Способность молекулярного кариотипирования «Оптима» определить, относится ли результат к плоду или к матери, существенно снижает риск получения ложных результатов.
Часто задаваемые вопросы
Что можно выявить при помощи теста «Оптима» при невынашивании беременности?
Перечень делеций и дупликаций, которые всегда отражаются в отчете:
Что нельзя обнаружить с помощью теста «Оптима»
Несмотря на то, что с помощью теста «Оптима» можно выявить множество хромосомных отклонений, его нельзя использовать для обнаружения сбалансированных хромосомных перестроек (инверсии, реципрокные транслокации). При этих перестройках сохраняется нормальное количество хромосомного материала, но нарушается его конфигурация. Изменения, как правило, не вызывают у носителя «перестройки» проблем со здоровьем, но могут стать причиной потери беременности или хромосомных нарушений у детей. Приблизительно каждый 500-й человек является носителем сбалансированной хромосомной перестройки.
Как можно снизить уровень контаминации материнскими клетками?
Сортировка плодного материала с целью отделения тканей плода для тестирования должна быть произведена еще до отправки образца. Ткань эмбриона обычно составляет менее 1% от общего объема плодного материала. Даже при тщательной сортировке образцов продуктов оплодотворения нет гарантии нахождения в них тканей эмбриона. Некоторые исследования показали, что более половины всех нормальных результатов тестирования образцов материала после спонтанного аборта являются таковыми из-за контаминации материнскими клетками
Как другие технологии тестирования могут повлиять на результаты теста?
Только тестирование «Оптима», основанное на анализе однонуклеотидных полиморфизмов, вместе с улучшенным биоинформатическим алгоритмом способно давать высокоточные результаты чаще и быстрее, чем стандартное кариотипирование. Так как для проведения данного тестирования не нужно выращивать клеточную культуру, вероятность отсутствия результата составляет менее 1%. Результаты тестирования предоставляются уже в течение 7 рабочих дней.
Как тестирование продуктов оплодотворения от лаборатории «Геномед» может помочь при подозрении на пузырный занос?
Пузырный занос может представлять собой серьезный риск для жизни и здоровья матери. Выявление пузырного заноса — критически важный фактор для лечения пациентки. В европейской популяции встречается с частотой 1/1000.


